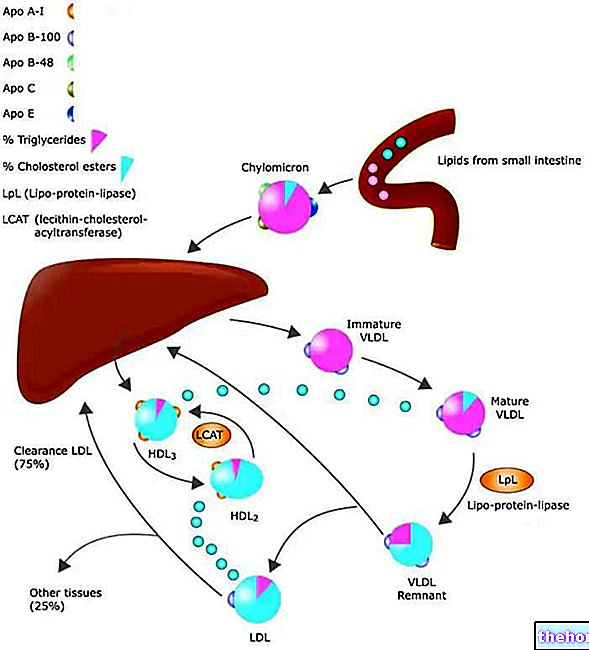
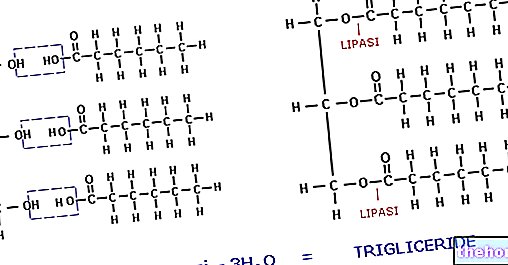
Lipoprotein lipase (LPL) er et enzym, der findes i endotelceller, der beklæder den indre overflade af blodkapillærer. Det er særligt koncentreret på niveauet af kapillært endotel af skeletmuskulaturvæv, hjertevæv og fedtvæv. Ikke overraskende er lipoproteinlipasens funktion at hydrolysere triglyceriderne i lipoproteinerne (chylomicroner og VLDL) og frigive to fedtsyrer gratis og en monoacylglycerol. Produkterne fra denne hydrolyse af triglycerider diffunderer ind i cellerne, hvor de i det væsentlige kan møde to skæbner: den første skal metaboliseres i skeletmuskulaturen og i hjertet, den anden, typisk for perioder med overfodring (energioverskud), skal bruges som substrater til resyntesen af triglycerider og derefter akkumuleres som en energireserve.
Insulin øger ekspressionen af lipoproteinlipase på niveau med hvidt fedtvæv, hvilket favoriserer hydrolyse af blodtriglycerider til glycerol og fedtsyrer; dermed kan sidstnævnte trænge ind i adipocytterne og derefter genestres med glycerol og danne reserve triglycerider.
Familiær lipoprotein lipase mangel (Burger-Grutz sygdom eller familiær hyperlipoproteinæmi type I)
Autosomal recessiv sygdom, med en forekomst svarende til et tilfælde hos 100.000 mennesker. Det forekommer hos emner, der er homozygote for en mutation på genet, der koder for lipoproteinlipase. Den deraf følgende mangel på dette enzym får dem, der er ramt af denne sygdom, til at vise særligt høje niveauer af triglycerider (normalt over 800-1000 mg / dL) på grund af blokering af metabolismen af chylomikroner. Alvorlig hypertriglyceridæmi ledsages siden barndommen af en større forekomst af pancreatitis, mavesmerter, eruptive xanthomer (gullige papler med røde konturer fordelt i kroppens områder udsat for pres) og hepatosplenomegali (unormal forstørrelse af lever og milt). kardiovaskulær risiko, mens retinopati undertiden er til stede.
Familiær APO-C2-mangel
Et meget vigtigt protein, fordi det er i stand til at aktivere lipoproteinlipasen, er Apo-lipo-protein-C2 eller APO-C2. En mangel på dette protein, udtrykt på overfladen af VLDL og chylomicrons, kan forårsage hyperlipoproteinæmi karakteriseret ved hypertriglyceridæmi (forhøjet triglycerider i blodet). APO-C2-mangel er derfor forbundet med en øget risiko for tidlig åreforkalkning og pancreatitis, mere almindelig hos gamle alder. Også i dette tilfælde er sygdommen forbundet med en autosomal recessiv mutation, specifikt i genet, der koder for APO-C2.
Lipoproteinlipaser, kost, medicin og kosttilskud
Lipoproteinlipase eller APO-C2-mangel kan behandles med en fedtfattig kost, der indtages i mængder, der ikke overstiger 10-20 gram om dagen. Mellemkædefedt, der binder direkte til albumin og ikke udnytter chylomikroner, der skal transporteres i blodbanen, bør klart foretrækkes.Samtidig er det nødvendigt at afskaffe alkohol og sikre en tilstrækkelig tilførsel af fedtopløselige vitaminer og fedtsyrer vigtig. Omega-3'erne af fiskeoprindelse (EPA og DHA) har især vist bemærkelsesværdige hypotiglyceridsænkende egenskaber og bruges som sådan i høje doser til at sænke triglyceridniveauer. Andre lægemidler med lignende aktivitet er fibrater og nikotinsyre, som udøver deres hypotryglyceridsænkende virkning også ved at øge ekspressionen af enzymet lipoproteinlipase.