Almindelighed
Wilsons sygdom, også kaldet hepatolentikulær degeneration, er en sjælden genetisk lidelse karakteriseret ved en ophobning af kobber i væv og organer i kroppen.

Det er en dødelig sygdom, og derfor er der behov for en terapeutisk behandling, der fjerner kobber fra vævene og forhindrer dets ophobning.
Det er Wilsons sygdom også
Wilsons sygdom, også kendt som hepatolentikulær degeneration, er en arvelig genetisk sygdom, der resulterer i en overdreven ophobning af kobber i visse organer og væv.
Det er en sjælden sygdom, der rammer 1 ud af 30.000 mennesker.
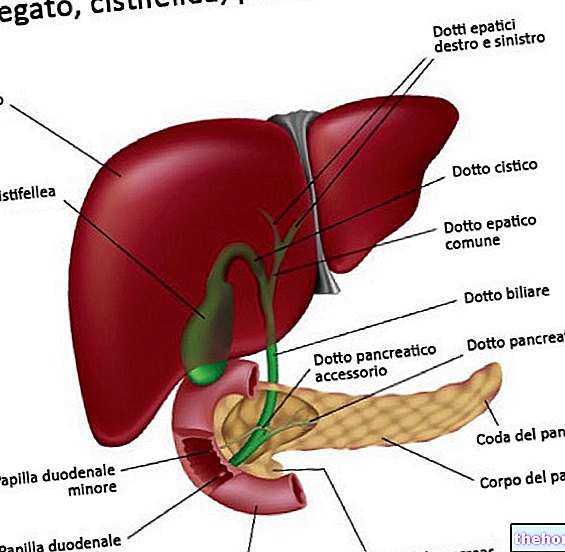
Akkumulering af kobber skyldes en defekt i dets stofskifte. Faktisk udskilles kobberet, der absorberes med kosten, ikke tilstrækkeligt, derfor forbliver det i kroppen og deponeres hovedsageligt i:
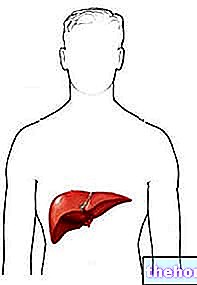
- lever;
- hjerne.
Og i mindre grad også i:
- hornhinde;
- nyrer;
- andre stoffer.
For store mængder kobber i disse områder forårsager skader på cellerne. De mest alvorlige virkninger findes i leveren og hjernen. I hjernen er det den lentikulære kerne, der lider de største konsekvenser: derfor det alternative navn på hepatolentikulær degeneration.
Årsager
Årsagen til Wilsons sygdom er en "ændring af ATP7B -genet, der er placeret på kromosom 13, som ikke længere udfører sin normale funktion.
ATP7B -genet har til opgave at fremme udskillelse, gennem galden, af overskydende kobber indeholdt i celler. Når ATP7B fejler, opbygges kobber i så massive doser, at det undslipper fra cellerne og strømmer ind i blodet. Blod derfor, kobber når kroppens forskellige væv.
Patogenese
Kobber indtages i kosten. Dens absorption sker i tarmen: her binder det sig til albumin (et plasmaprotein) og når leveren. På dette tidspunkt:
hos en sund person:
- ATP7B fremmer bindingen mellem kobber og ceruloplasmin. Ceruloplasmin er et plasmaprotein, der bruges til transport og udskillelse af kobber.
Hos en person med Wilsons sygdom dog:
- ATP7B virker ikke. Derfor favoriserer det ikke bindingen mellem kobber og ceruloplasmin.
- Kobber forbliver bundet til albumin, udskilles ikke og ophobes i leverceller.
- Levercellerne mætter enhver kobberlagerkapacitet i dem.
- Kobber-albuminkomplekset er derfor i overskud. Derfor undslipper den fra hepatocytterne og kommer ind i blodet.
- Gennem blodet når kobber kroppens andre væv.
Det første organ til at betale konsekvenserne er derfor leveren; hjernen, nyrerne og hornhinden følger.

Hvorfor spredes kobber til tekstiler?
Hos personer med Wilsons sygdom cirkulerer kobber i blodet bundet til albumin. Kobber-albuminbindingen er meget mere labil end den mellem kobber og ceruloplasmin. Faktisk er der ringe affinitet mellem de to første c ". Når kobberet komplekseret med albumin når vævene og de forskellige organer, støder det på stoffer, som det har større affinitet til og binder sig til. Konsekvenserne er to:
- Væv og organer er beriget med kobber.
- Koncentrationen af kobber (cupremia) i blodet falder.

Fra Wikipedia - Mor og far har begge en muteret allel. På grund af denne allels recessive karakter manifesterer de ikke nogen sygdom, men er sunde bærere. Begge forældre kan videregive en muteret allel til et barn hver. I dette tilfælde vil barnet være homozygotisk for den givne allel og vil manifestere sygdommen. I alle andre tilfælde forårsager tilstedeværelsen af en eller begge sunde alleler ingen forstyrrelser.
Arv
Wilsons sygdom er en autosomal recessiv arvelig sygdom.
- Autosomal, fordi ATP7B-genet er placeret på kromosom 13, et ikke-kønskromosom.
- Recessiv, fordi den muterede allel, som bestemmer sygdommen, er recessiv i forhold til den raske. For at blive syg skal et individ have begge muterede alleler. Faktisk er en enkelt muteret allel ikke nok til at forårsage sygdommen. Én ud af 100 mennesker omkring bærer en ændret ATP7B -allel Figuren forklarer klart dette koncept.
Symptomer
Yderligere oplysninger: Wilsons sygdomssymptomer
Selvom det er en arvelig genetisk sygdom, er der i de allerførste år ingen forstyrrelser. De første symptomer, baseret på leveren, vises omkring 6 år. Dette er normalt den mindste tid, det tager for kobber at ophobe sig i skadelige mængder. I nogle tilfælde kan begyndelsen forekomme i slutningen af ungdomsårene eller endda omkring 30-40 års alderen. Over tid forekommer der også lidelser i andre væv.
Lever symptomer
Leveren er det første organ, der er berørt, fordi det er det første distrikt, hvor kobberet, der absorberes fra kosten, ankommer. Leversundhed forværres gradvist. Evolution begynder typisk i ungdomsårene og følger følgende forløb:
- Hepatitis.
- Ikke alvorlig skrumpelever.
- Alvorlig skrumpelever.
En tilstand defineret af lægen er skabt med udtrykket leversvigt: leveren er ikke længere i stand til at udføre sine funktioner.
Typiske tegn på leversvigt er:
- Gulsot.
- Mavesmerter.
- Han trak sig tilbage.
- Udvidelse af leveren (hepatomegali)
- Udvidelse af milten (splenomegali)
Hjernesymptomatologi
Kobber når først hjernen, når leveren ikke længere kan holde den begrænset til sine egne celler.
Indskud i hjernen forårsager neurologisk skade af en anden art:
- Fysiske lidelser.
- Rystelser i lemmerne.
- Langsom bevægelse.
- Problemer med tale (dysartri).
- Problemer med at skrive.
- Synkebesvær (dysfagi).
- Ustabilitet i gang.
- Migræne.
- Epilepsi.
- Muskelsvaghed og stivhed.
- Adfærdsforstyrrelser.
- Humør ændrer sig.
- Depression.
- Manglende evne til at koncentrere sig.
- Personligheden ændrer sig.
- Demens.
Hvis patienten ikke behandles, forværres den neurologiske skade mere og mere: individet bliver helt afhængig af andre, for at fodre og bevæge sig.
Andre stoffer

Endvidere kan kobber også aflejres i nyrerne. Nyreskader opstår, hvilket resulterer i:
- Aminosyre. Tilstedeværelse af aminosyrer i urinen.
- Glykosuri. Tilstedeværelse af glukose i urinen.
- Phosphaturia. Tilstedeværelse af fosfor i urinen
- Uricosuria. Tilstedeværelse af urinsyre i urinen.
- Calciuria. Tilstedeværelse af calcium i urinen.
Under normale forhold vil alle disse tabte stoffer blive absorberet igen. Derfor ændrer nyreakkumuleringen af kobber strukturen og reabsorptionen af stoffer, der stadig er nyttige for organismen.
Andre mulige symptomer på Wilsons sygdom er:
- Anæmi.
- Pankreatitis.
- Menstruationsproblemer.
- Abort.
- For tidlig osteoporose.
Diagnose
Hvis mistanke om Wilsons sygdom er nyttige diagnostiske tests:
- Blodprøver, for at teste:
- Koncentrationer af ceruloplasmin. Et lavt niveau, under 20 mg / 100 ml, er tegn på sygdommen. Den normale værdi er 30 mg / 100 ml.
- Koncentrationen af kobber (cupremia). Hvis det er mindre end normalt, er det tegn på sygdommen.
- Mulig hæmolytisk anæmi.
- Lever- og nyrefunktioner gennem deres respektive markører (transaminaser, azotæmi osv.)
- Urinalyse for at evaluere mængden af tilstedeværende kobber (cupruria). Niveauer over det normale er tegn på sygdommen. Typisk udskiller mennesker med tilstanden omkring 100μg kobber i deres urin hver 24. time.
- Optometrisk undersøgelse for at påvise tilstedeværelsen af Kaiser-Fleischer-ringen.
- Leverbiopsi, til måling af kobberindholdet i leverceller. Det patologiske niveau af kobber er over 100μg pr. Gram lever. Det er også nyttigt til vurdering af cirrose.
- En MR i hjernen for at evaluere sundheden for den lentikulære kerne, som vi husker er hjernens område påvirket af akkumulering af kobber.
- En genetisk DNA -test.
Den samtidige tilstedeværelse af:
- Kaiser-Fleischer ring.
- Tegn på levercirrhose.
- Læsion af den lentikulære kerne.
efterlader ingen tvivl om diagnosen.
Målt parameter
Cupræmi
110 μg / ml
<100μg / ml
Cupruria
100 μg / 24 timer
>> 100 μg / 24 timer
Ceruloplasmin
30 mg / ml
<20 mg / ml
Terapi
Se også: Lægemidler til binyrebarkinsufficiens
Ubehandlet er Wilsons sygdom dødelig. Døden kan opstå selv et par år efter, at de første symptomer opstår. Patienten bliver gradvist forværret af sin tilstand, bliver i stigende grad afhængig af andre, og i mangel af specifik behandling kan lever- og hjerneskade være irreversibel.
Terapien består af:
- Reducer kobberaflejringer i leveren.
- Kontroller absorptionen af kobber i tarmen.
- Reducer indførelsen af kobber taget med kosten.
- Levertransplantation.
Reducer kobberaflejringer
Det er det vigtigste trin i at redde patientens liv. Det er baseret på administration af:
- Penicillamin.
- Trientina.
Penicillamin er det foretrukne lægemiddel, dets administration foregår oralt og skal tages for livet.Det repræsenterer et chelateringsmiddel, der er i stand til at udskille overskydende kobber og føre det til nyrerne for udskillelse. Det kan dog forårsage uønskede virkninger i selve nyren. I disse tilfælde er det tilrådeligt at afbryde behandlingen for at løse de opståede problemer og at vedtage et alternativ baseret på trientin.
Trientin er også et chelateringsmiddel. Det administreres oralt og virker som penicillamin. Det er ikke så effektivt, men bivirkningerne er også mindre.
Kontroller intestinal absorption af kobber
Det er muligt at reducere absorptionen af kobber ved at tage zink, hvilket forhindrer ophobning af kobber i leveren. Administration af zink anbefales, når Wilsons sygdom er i sine tidlige stadier. Med andre ord, når kobberet endnu ikke har invaderet de andre væv. Terapi er effektiv, når den kombineres med penicillaminbehandling.
Reducer indførelsen af kobber
Forbruget af visse fødevarer, der er rige på kobber, bør begrænses, såsom:
- Valnødder.
- Lever.
- Svampe.
- Chokolade.
- Fisk og skaldyr.
Samlet set bør det daglige indtag af kobber ikke overstige 2 mg.
Levertransplantation
Dette er den nødvendige terapi, hvis:
- Leverskade er irreversibel. I dette tilfælde taler vi om alvorlig cirrose.
- Tidligere behandlinger har været ineffektive.
Prognose
Jo tidligere behandling påbegyndes, jo bedre bliver prognosen og livskvaliteten.
At intervenere sent betyder begrænsning og kun delvis forbedring af lever- og hjerneskade på grund af overskydende kobber. Nogle funktioner forbliver faktisk uopretteligt kompromitterede.
I alvorlige tilfælde er den eneste løsning for en bedre prognose levertransplantation.