Aktive ingredienser: Filgrastim
Nivestim 12 MU / 0,2 ml injektions- / infusionsvæske, opløsning
Nivestim 30 MU / 0,5 ml injektions- / infusionsvæske, opløsning
Nivestim 48 MU / 0,5 ml injektions- / infusionsvæske, opløsning
Hvorfor bruges Nivestim? Hvad er det for?
Hvad er Nivestim
Nivestim indeholder det aktive stof filgrastim, der tilhører en gruppe proteiner kaldet cytokiner og ligner meget et naturligt protein (granulocytkolonistimulerende faktor [G-CSF]) produceret af menneskekroppen. Filgrastim stimulerer knoglemarven (det væv, der danner nye blodlegemer) til at producere flere blodlegemer, især nogle typer hvide blodlegemer. hvide blodlegemer er vigtige, fordi de hjælper kroppen med at bekæmpe infektioner.
Hvad Nivestim bruges til
Din læge har ordineret Nivestim til dig for at hjælpe din krop med at lave flere hvide blodlegemer. Din læge vil forklare, hvorfor du er ordineret Nivestim. Nivestim er nyttig i en række forskellige kliniske tilstande, såsom:
- kemoterapi
- knoglemarvstransplantation,
- alvorlig kronisk neutropeni (neutropeni er et unormalt lavt antal af en bestemt type hvide blodlegemer, også kendt som neutrofiler),
- neutropeni hos patienter med hiv -infektion,
- mobilisering af perifere blodstamceller.
Kontraindikationer Når Nivestim ikke bør bruges
Brug ikke Nivestim
- hvis du er allergisk over for filgrastim eller et af de øvrige indholdsstoffer i dette lægemiddel.
Forholdsregler ved brug Det, du skal vide, før du tager Nivestim
Tal med din læge, apotek eller sygeplejerske, før du tager Nivestim: - hvis du har en anden sygdom (især hvis du tror, du har en "infektion),
- hvis du har hoste, feber og vejrtrækningsbesvær. De kan være sekundære for lungeproblemer (se også afsnit 4 "MULIGE ADVERSE Hændelser"),
- hvis du har seglcelleanæmi (en arvelig blodsygdom, der påvirker røde blodlegemer),
- hvis du har ondt i maven i øverste venstre eller hvis du har smerter i skulderen. Det kan være en konsekvens af en miltsygdom (se afsnit 4 "MULIGE ADVERSE Hændelser"),
- hvis du lider af specifikke blodsygdomme (f.eks. Kostmanns syndrom, myelodysplastisk syndrom, forskellige typer leukæmi)
- hvis du lider af knogleskørhed. Din læge kan kontrollere densiteten af dine knogler regelmæssigt.
Hvis du har brug for en knoglescanning, skal du fortælle det til din læge eller sygeplejerske, at du er i behandling med Nivestim.
Fortæl det straks til din læge eller sygeplejerske, hvis du oplever pludselige tegn på allergi såsom udslæt, kløe eller nældefeber på huden, hævelse af ansigt, læber, tunge eller andre dele af kroppen, åndenød, hvæsen eller vejrtrækningsbesvær under behandlingen med Nivestim, fordi disse kan være tegn på en alvorlig allergisk reaktion.
Mens du bliver behandlet med Nivestim, kan du have brug for regelmæssige blodprøver for at kontrollere antallet af neutrofiler og andre hvide blodlegemer i dit blod. Med disse data bestemmer lægen effektiviteten af behandlingen, og om den skal fortsætte.
Tab af reaktion på filgrastim
Hvis du har nedsat respons eller manglende reaktion på behandling med filgrastim, vil din læge undersøge årsagerne, herunder muligheden for at du har udviklet antistoffer, der neutraliserer filgrastims aktivitet.
Interaktioner Hvilke lægemidler eller fødevarer kan ændre virkningen af Nivestim
Du må ikke behandles med Nivestim i 24 timer før og 24 timer efter behandling med kemoterapi.
Fortæl det til din læge eller apotek, hvis du tager anden medicin eller har brugt det for nylig.
Advarsler Det er vigtigt at vide, at:
Graviditet og amning
Spørg din læge eller apotek til råds, før du tager medicin.
Filgrastim er ikke undersøgt hos gravide. Det er vigtigt, at du fortæller din læge, hvis du er gravid, hvis du tror, at du kan blive gravid, eller hvis du planlægger at blive gravid, da din læge kan beslutte, at du ikke kan bruge denne medicin. Filgrastim kan have negative virkninger på din evnen til at blive gravid eller føre til afslutning af en graviditet.
Det vides ikke, om filgrastim går over i modermælk. Derfor kan din læge beslutte, at du ikke kan bruge denne medicin, hvis du ammer.
Kørsel og brug af maskiner
Filgrastim har beskedne virkninger på evnen til at føre motorkøretøj og betjene maskiner.Hvis du føler dig træt, skal du være forsigtig, når du kører bil eller betjener maskiner.
Nivestim indeholder sorbitol
Denne medicin indeholder sorbitol (E420). Hvis din læge har fortalt dig, at du ikke tåler nogle sukkerarter (fruktose), skal du kontakte din læge, inden du tager dette lægemiddel. Denne medicin indeholder også natrium i mindre end 1 mmol (23 mg) pr. Dosis og er derfor i det væsentlige "natriumfri".
Dosis, metode og administrationstidspunkt Sådan bruges Nivestim: Dosering
Brug altid dette lægemiddel nøjagtigt efter lægens anvisning. Spørg din læge, hvis du er i tvivl.
Denne medicin gives ved injektion, enten ved intravenøs infusion (drop) eller subkutant i vævet, der er placeret direkte under huden.
Hvis du bruger dette lægemiddel ved subkutan injektion, kan din læge finde det hensigtsmæssigt, at du selv injicerer det. Din læge eller sygeplejerske vil vise dig, hvordan du injicerer (instruktioner om selvinjektion findes i slutningen af denne indlægsseddel). Prøv ikke at injicere dig selv, medmindre du er blevet oplært til det. Nogle oplysninger, du har brug for, er rapporteret nederst i denne indlægsseddel er det imidlertid nødvendigt med et omhyggeligt og konstant samarbejde med din læge for den korrekte behandling af din sygdom.
Mængden af Nivestim, du har brug for, afhænger af den sygdom, du tager Nivestim for, og din kropsvægt.
Nivestim og kemoterapi-associeret neutropeni
Den sædvanlige dosis til voksne og børn er 0,5 millioner enheder (5 mikrogram) pr. Kg legemsvægt pr. Dag. For eksempel, hvis du vejer 60 kg, er din daglige dosis 30 millioner enheder (300 mikrogram). Behandlingen kan vare op til 14 dage. For nogle sygdomme kan forlænget behandling i op til ca. en måned være påkrævet.
Nivestim og knoglemarvstransplantation
Den sædvanlige startdosis er 1 million enheder (10 mikrogram) pr. Kg legemsvægt pr. Dag ved infusion. Hvis du f.eks. Vejer 60 kg, er din daglige dosis 60 millioner enheder (600 mikrogram). Du vil generelt modtage den første dosis mindst 24 timer efter kemoterapi, men inden for 24 timer efter en knoglemarvstransplantation. Lægen vil derefter foretage blodprøver for at kontrollere effekten af behandlingen og fastslå dens varighed.
Nivestim og alvorlig kronisk neutropeni
Den sædvanlige startdosis er mellem 0,5 millioner (5 mikrogram) og 1,2 millioner (12 mikrogram) enheder pr. Kilogram legemsvægt pr. Dag, som en enkelt eller delt dosis. Din læge vil derefter foretage blodprøver for at kontrollere effekten af behandlingen og bestemme den mest passende dosis for dig.I tilfælde af neutropeni er det nødvendigt med langvarig behandling med Nivestim.
Nivestim og neutropeni hos patienter med hiv -infektion
Den sædvanlige startdosis er mellem 0,1 (1 mikrogram) og 0,4 millioner enheder (4 mikrogram) pr. Kg legemsvægt pr. Dag. Din læge vil foretage blodprøver med jævne mellemrum for at kontrollere effekten af behandlingen. Når antallet af hvide blodlegemer er vendt tilbage til det normale, kan hyppigheden af dosering reduceres til mindre end en gang om dagen. Opretholdelse af normale antal hvide blodlegemer kan kræver langvarig behandling med Nivestim.
Nivestim og perifer blod stamcelletransplantation
Hvis du donerer stamceller til dig selv, er den sædvanlige dosis 0,5 millioner (5 mikrogram) til 1 million enheder (10 mikrogram) pr. Kilogram legemsvægt pr. Dag. Behandling med Nivestim varer op til 2 uger. Lægen vil foretage blodprøver for at bestemme det bedste tidspunkt at indsamle stamcellerne.
Hvis du donerer stamceller til en anden person, er den sædvanlige dosis 1 million enheder pr. Kilogram kropsvægt pr. Dag. Behandling med Nivestim varer i 4 til 5 dage.
Hvis du har glemt at bruge Nivestim
Hvis du har glemt at injicere en dosis, skal du kontakte din læge eller apotek og spørge dem, hvornår den næste dosis skal injiceres. Brug ikke en dobbeltdosis til at kompensere for en glemt injektion.
Sådan slutter Nivestim -behandlingen
Din læge vil fortælle dig, hvornår du skal stoppe med at bruge Nivestim. Det er helt normalt at have flere behandlingsforløb med Nivestim.
Spørg din læge eller apotek, hvis du har yderligere spørgsmål om brugen af medicinen.
Overdosering Hvad skal man gøre, hvis man har taget for meget Nivestim
Hvis du har brugt mere Nivestim, end du burde, skal du kontakte din læge eller apotek så hurtigt som muligt.
Bivirkninger Hvad er bivirkningerne af Nivestim
Ligesom al anden medicin kan denne medicin forårsage bivirkninger, men ikke alle får bivirkninger.
Allergiske reaktioner på filgrastim, herunder udslæt, hævede kløende hudområder og anafylaksi (svaghed, blodtryksfald, vejrtrækningsbesvær og synke og hævelse af ansigtet) er blevet rapporteret. Hvis du tror, du har denne type reaktioner, skal du stoppe med at tage Nivestim -injektioner og straks søge lægehjælp.
Øget størrelse på milten og meget sjældne tilfælde af brud på milten er blevet rapporteret. Nogle tilfælde af bristet milt har været dødelige. Det er vigtigt, at du straks kontakter din læge, hvis du oplever smerter i venstre øvre del af maven eller skulderbladet, da dette kan være et tegn på problemer med milten.
Fortæl det straks til din læge, hvis du oplever en eller flere af følgende bivirkninger under behandlingen:
- hævelse eller hævelse, som kan være relateret til nedsat vandladning, vejrtrækningsbesvær, oppustethed og mæthed og en generel træthedsfølelse. Disse symptomer kommer normalt hurtigt.
Disse kan være symptomer på en ualmindelig sygdom (kan ramme op til 1 ud af 100 mennesker) kendt som "kapillær lækagesyndrom", som får blod til at sive ind i kroppen fra små blodkar, og som kræver akut lægehjælp.
Det er også meget vigtigt at fortælle det til din læge, hvis du tror, du har en "infektion. Der er mange måder, hvorpå en" infektion kan manifestere sig. Du bør kontrollere, at temperaturen ikke er og ikke overstiger 37,8 ° C, kuldegysninger eller andre tegn på infektion, såsom rødme i huden, ondt i halsen, diarré, øre smerter, åndedrætsbesvær eller smerter som hoste og astma Disse symptomer kan skyldes alvorlige lungebivirkninger, såsom lungebetændelse og respiratorisk nødsyndrom hos voksne, som kan forårsage dødsfald Hvis du har feber eller andre symptomer, skal du straks kontakte din læge og gå direkte til et hospital.
Hvis du har seglcelleanæmi, skal du informere din læge, før du starter behandling med Nivestim. Der er rapporteret om kriser med seglcelleanæmi hos nogle patienter med denne sygdom behandlet med filgrastim.
Meget almindelige bivirkninger (rammer mere end 1 ud af 10 patienter)
- Følelse eller sygdom
- Smerter i knogler og muskler. Spørg din læge, hvilken medicin du skal tage for at lindre dette
- Næseblod
- Fald i blodglukoseniveauer, som kan få dig til at føle dig sulten, syg, svag, træt, rystet eller forvirret eller forårsage svedtendens, hovedpine, sløret syn eller øget puls
- Stigning i leverenzymværdier eller ændringer i blodprøver. Din læge vil foretage blodprøver for at kontrollere dette
- Øget urinsyre, der kan manifestere sig med urinsyregigt
- Brystsmerter
Almindelige bivirkninger (rammer op til 1 ud af 10 patienter)
- Svaghed
- Generaliseret træthed
- Hovedpine
- Forstoppelse eller diarré
- Mistet appetiten
- Betændelse og sårdannelse i munden og tarmens indre foring
- Hoste
- Ondt i halsen
- Hårtab
- Udslæt
- Forstørret lever
- Udtynding af knoglerne
- Smerter på injektionsstedet
- Betændelse i blodkarrene
- Fald i blodplader (celler involveret i koagulation) - med øget risiko for blødning eller blå mærker
Ikke almindelige bivirkninger (rammer op til 1 ud af 100 patienter)
- Uspecificeret smerte
- Tilstedeværelse af blod eller protein i urinen
Sjældne bivirkninger (forekommer hos op til 1 ud af 1.000 patienter)
- Leverskade forårsaget af blokering af de små vener i leveren (veno-okklusiv sygdom)
- En ændring i reguleringen af væsker i kroppen, som kan føre til hævelse
Meget sjældne bivirkninger (forekommer hos op til 1 ud af 10.000 patienter)
- Unormal røntgenradiografi af lungerne (lungeinfiltration)
- Violetfarvede, hævede og smertefulde læsioner på lemmerne og undertiden i ansigt og hals, forbundet med feber (Sweet's syndrom)
- Betændelse i hudens blodkar (kutan vaskulitis) • Forværring af reumatoid arthritis findes allerede
- Usædvanlig ændring i urinen
Frekvens ikke kendt (frekvens kan ikke estimeres ud fra de tilgængelige data)
- Hævelse og smerter i leddene, som ved gigt (pseudogout).
Graft versus host -sygdom (GvHD) kan forekomme hos patienter, der gennemgår stamcelledonation eller knoglemarvstransplantation - dette er en reaktion fra donorcellerne mod den person, der modtager transplantationen; tegn og symptomer på, at de omfatter udslæt på håndflader eller såler og sår og sår i mund, tarm, lever, hud eller øjne, lunger, skede og led. Nogle tilfælde af GvHD har været dødelige.
Bivirkninger, du kan opleve, hvis du er en stamceldonor for en anden person, er:
Meget almindelige bivirkninger (rammer mere end 1 ud af 10 patienter)
- Hovedpine
- Smerter i knoglerne eller musklerne. Spørg din læge, hvilken medicin du skal tage for at lindre dette
- Ændringer i hvide blodlegemer eller blodplader (din læge vil kontrollere dem med blodprøver)
Almindelige bivirkninger (rammer op til 1 ud af 10 patienter)
- Forøgelse af niveauerne af nogle leverenzymer (din læge vil overvåge dem)
Ikke almindelige bivirkninger (rammer op til 1 ud af 100 patienter)
- Alvorlig allergisk reaktion
- Problemer med milten
- Øgede niveauer af urinsyre, der kan forekomme med gigt
- Forværring af leddegigt
Indberetning af bivirkninger
Tal med din læge eller apotek eller sygeplejerske, hvis du får bivirkninger. Dette inkluderer eventuelle bivirkninger, der ikke er anført i denne indlægsseddel. Du kan også rapportere bivirkninger direkte via det nationale rapporteringssystem anført i tillæg V.
Udløb og opbevaring
Opbevar denne medicin utilgængeligt for børn.
Brug ikke dette lægemiddel efter den udløbsdato, der står på den ydre karton og på sprøjtemærket efter forkortelsen EXP / EXP. Udløbsdatoen refererer til den sidste dag i måneden.
Opbevares og transporteres køligt (2 ° C - 8 ° C). Må ikke fryses. Opbevar den fyldte sprøjte i sin originale emballage for at beskytte medicinen mod lys.
Sprøjten kan opbevares uden for køleskabet og efterlades ved stuetemperatur i en enkelt periode på op til 7 dage (dog ikke over 25 ° C).
Brug ikke Nivestim, hvis du bemærker, at det er grumset, eller der er partikler i det.
Smid ikke medicin ned i spildevand eller husholdningsaffald. Spørg din apotek om, hvordan du skal smide medicin, du ikke længere bruger. Dette vil hjælpe med at beskytte miljøet.
Andre oplysninger
Hvad Nivestim indeholder
- Den aktive ingrediens er filgrastim. Hver milliliter indeholder 60 millioner enheder [MU] (600 mikrogram) eller 96 millioner enheder [MU] (960 mikrogram) filgrastim.
- Nivestim 12 MU / 0,2 ml injektions- / infusionsvæske, opløsning: hver fyldt injektionssprøjte indeholder 12 millioner enheder (MU), 120 mikrogram filgrastim i 0,2 ml (svarende til 0,6 mg / ml).
- Nivestim 30 MU / 0,5 ml injektions- / infusionsvæske, opløsning: Hver fyldt injektionssprøjte indeholder 30 millioner enheder (MU), 300 mikrogram filgrastim i 0,5 ml (svarende til 0,6 mg / ml).
- Nivestim 48 MU / 0,5 ml injektions- / infusionsvæske, opløsning: Hver fyldt injektionssprøjte indeholder 48 millioner enheder (MU), 480 mikrogram filgrastim i 0,5 ml (svarende til 0,96 mg / ml).
- Øvrige indholdsstoffer er eddikesyre, natriumhydroxid, sorbitol E420, polysorbat 80 og vand til injektionsvæsker.
Hvordan Nivestim ser ud og pakningens indhold
Nivestim er en klar, farveløs opløsning til injektion / infusion i en fyldt injektionssprøjte med en beskyttende (rustfri) injektionskanyl. Der kan være 1, 5, 8 eller 10 sprøjter i hver pakning.
Patientinstruktioner til selvinjektion
Dette afsnit indeholder oplysninger om, hvordan du selv injicerer Nivestim. Det er vigtigt, at du ikke selv prøver at injicere medicinen, før du er specielt uddannet af din læge eller sygeplejerske.
Det er også vigtigt, at du smider sprøjten i en kanyleprikket beholder. Spørg din læge eller sygeplejerske, hvis du har spørgsmål eller bekymringer om selvinjektion.
Hvordan injicerer jeg mig selv?
Normalt gives Nivestim to gange dagligt ved injektion i vævene placeret under huden, også kendt som en subkutan injektion.
At lære selvindsprøjtningsproceduren betyder at undgå at vente hjemme på, at sygeplejersken ankommer, langt mindre at skulle gå til hospitalet eller klinikken hver dag for at modtage injektionen.
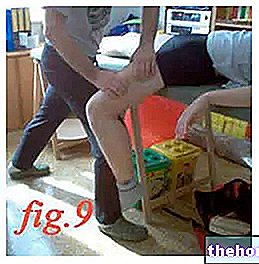
Du bliver nødt til at injicere dig selv på samme tid hver dag. De mest egnede steder at injicere er:
- forsiden af låret,
- maven, bortset fra området omkring navlen.
Det er bedst at altid skifte injektionssted hver dag for at undgå smerter i et bestemt område.
Udstyr nødvendigt til administration
Følgende udstyr er påkrævet, når der udføres en "subkutan selvinjektion:"
- En ny Nivestim fyldt injektionssprøjte.
- En skarp beholder (kanylepunktur) til sikker bortskaffelse af brugte sprøjter.
- Antiseptiske servietter (hvis anbefalet af din læge eller sygeplejerske).
Hvordan giver jeg Nivestim subkutan selvinjektion?
- Prøv at injicere dig selv på omtrent samme tidspunkt hver dag.
- Tag Nivestim-sprøjten ud af køleskabet, og lad den nå stuetemperatur (ca. 25 ° C). Det tager 15-30 minutter. Kontroller datoen på pakken for at sikre, at medicinen ikke er udløbet. Hav beholderen til opsamling af skarpe affaldsmaterialer i nærheden.
- Se efter et godt oplyst arbejdssted for din injektion og for at kontrollere din foreskrevne dosis.
- Vask dine hænder grundigt med sæbe og vand.
- Fjern sprøjten fra blisterpakningen, og kontroller, at opløsningen, den indeholder, er klar, farveløs og praktisk talt fri for synlige partikler. Brug ikke Nivestim -sprøjten, hvis væsken har flydende partikler i den, eller hvis noget af væsken er lækket ud af sprøjten.
- Hold sprøjten med kanylen pegende opad. Fjern beskyttelseshætten fra kanylen. Sprøjten er nu klar til brug. Der kan være en lille luftboble i sprøjten. Du behøver ikke at fjerne luftboblen før injektion. Det er ikke farligt at injicere opløsningen i nærværelse af en luftboble.
- Beslut, hvor du skal injicere Nivestim - find et sted på forsiden af maven eller på forsiden af lårene. Vælg et andet injektionssted hver gang. Vælg ikke et smertefuldt, rødt, forslået eller arret område. Hvis din sygeplejerske eller læge anbefaler det, skal du rengøre hudoverfladen med et desinfektionsmiddel.
- Tag en "bred hudfold mellem din tommelfinger og pegefinger, pas på ikke at røre ved det område, du lige har rengjort."
- Med den anden hånd skal du stikke nålen ind under huden i en vinkel på cirka 45 °.
- Træk let i stemplet for at kontrollere, at der ikke kommer blod ind i sprøjten. Hvis du ser blod i sprøjten, skal du trække nålen ud og indsætte den et andet sted. Skub langsomt ned på stemplet, indtil hele indholdet af sprøjten er tømt.
- Efter injektion af opløsningen fjernes nålen fra huden.
- Ved at følge instruktionerne herunder for den aktive eller passive nålebeskyttelsesenhed, skal du sørge for, at enheden dækker nålen.
- Returner sprøjten til beholderen til skarpe. Forsøg ikke at udskifte beskyttelseskappen.
- Opbevar brugte sprøjter utilgængeligt for børn.
- Smid ALDRIG sprøjter i din husholdningsaffaldsbeholder.
Husk
De fleste mennesker kan lære om subkutan selvinjektion, men hvis du har mange problemer, skal du ikke være bange for at spørge din læge eller sygeplejerske om hjælp og råd.
Brug af Active Ultrasafe nålebeskytter til Nivestim 12 MU / 0,2 ml injektions- / infusionsvæske, opløsning
Den fyldte injektionssprøjte er udstyret med en nålesikkerhedsanordning, UltraSafe Needle Guard, der beskytter mod utilsigtet nålestik.Hold dine hænder bag nålen, når du håndterer den fyldte sprøjte.
- Udfør injektionen ifølge den teknik, der er beskrevet ovenfor.
- Når du er færdig med din injektion, skal du skubbe nålebeskytteren fremad, indtil nålen er helt dækket (enheden klikker på plads).
Brug af Ultrasafe Passive Needle Guard til Nivestim 30 MU / 0,5 ml injektions- / infusionsvæske, opløsning og Nivestim 48 MU / 0,5 ml injektions- / infusionsvæske, opløsning
Den fyldte injektionssprøjte er udstyret med en kanylebeskyttelse, UltraSafe Needle Guard, der beskytter mod utilsigtet nålestik.Hold dine hænder bag nålen, når du håndterer den fyldte sprøjte.
- Udfør injektionen ifølge den teknik, der er beskrevet ovenfor.
- Mens du holder sprøjten med fingrene hvilende på støttekanten, skal du trykke på stemplet, indtil injektionen af hele dosis er fuldført. Det passive nålebeskyttelsessystem aktiveres IKKE, hvis FULD dosis ikke er blevet administreret.
- Fjern kanylen fra din hud, slip derefter stemplet, og sprøjten bevæger sig fremad, indtil afskærmningen har dækket nålen og klikker på plads.
FØLGENDE OPLYSNINGER ER KUN TILSÆTT TIL LÆGER ELLER SUNDHEDSPROFessionelle
Nivestim indeholder ingen konserveringsmidler. På grund af risikoen for bakteriel kontaminering er Nivestim sprøjter kun til engangsbrug.
Utilsigtet udsættelse for frysetemperaturer i op til 24 timer har ingen negativ indvirkning på Nivestims stabilitet.Den frosne fyldte injektionssprøjte kan optøs og returneres til køleskabet til senere brug. Hvis eksponeringen for lave temperaturer er større end 24 timer, eller hvis den er frosset mere end én gang, må Nivestim IKKE bruges mere.
Nivestim må ikke fortyndes i natriumchloridopløsning. Dette lægemiddel må ikke blandes med andre lægemidler end dem, der er beskrevet nedenfor. Hvis den ikke fortyndes som beskrevet nedenfor, kan fortyndet filgrastim absorbere til glas og plastmaterialer.
Om nødvendigt kan Nivestim fortyndes i 50 mg / ml (5%) glucoseopløsning til infusion. Fortynding til slutkoncentrationer <0,2 MU (2 mikrogram) pr. Ml anbefales aldrig. Opløsningen bør inspiceres visuelt før brug. Kun klare opløsninger uden synlige partikler bør anvendes. For patienter behandlet med filgrastim fortyndet til koncentrationer under 1,5 MU (15 mikrogram) pr. Ml, humant serumalbumin (HSA) til en slutkoncentration på 2 mg / ml.
Eksempel: I et sidste volumen, der skal injiceres på 20 ml, bør totale filgrastim -doser under 30 MU (300 mikrogram) administreres ved tilsætning af 0,2 ml af en 200 mg / ml (20%) human albuminopløsning. Fortyndet med 50 mg / ml (5%) glucoseopløsning til infusion, er Nivestim kompatibel med glas og forskellige plastmaterialer, såsom PVC, polyolefin (en copolymer af polypropylen og polyethylen) og polypropylen.
Efter fortynding: Kemisk og fysisk stabilitet i den fortyndede infusionsvæske, opløsning er påvist i 24 timer ved 2 ° C til 8 ° C. Fra et mikrobiologisk synspunkt bør produktet bruges med det samme. Hvis det ikke bruges med det samme, er opbevaringstider og -betingelser inden brug brugerens ansvar og vil normalt ikke overstige 24 timer ved 2 ° C til 8 ° C, medmindre fortynding har fundet sted under aseptisk kontrollerede og validerede forhold.
Indlægsseddel: AIFA (Italian Medicines Agency). Indhold offentliggjort i januar 2016. De foreliggende oplysninger er muligvis ikke opdaterede.
For at få adgang til den mest opdaterede version er det tilrådeligt at få adgang til webstedet AIFA (Italian Medicines Agency). Ansvarsfraskrivelse og nyttige oplysninger.
01.0 LÆGEMIDLETS NAVN
NIVESTIM 12 MU / 0,2 ML OPLØSNING TIL INJEKTION / INFUSION
02.0 KVALITATIV OG KVANTITATIV SAMMENSÆTNING
Hver ml injektions- eller infusionsvæske, opløsning indeholder 60 millioner enheder [MU] (600 mcg) filgrastim *.
Hver fyldt injektionssprøjte indeholder 12 millioner enheder (MU) (120 mcg) filgrastim i 0,2 ml (0,6 mg / ml).
* rekombinant methioningranulocytkolonistimulerende faktor [GCSF]) produceret i Escherichia Coli (BL21) med rekombinant DNA -teknologi.
Hjælpestof (er) med kendt virkning
Hver ml opløsning indeholder 50 mg sorbitol.
Den fulde liste over hjælpestoffer findes i afsnit 6.1.
03.0 LÆGEMIDDELFORM
Injektions- / infusionsvæske, opløsning.
Klar, farveløs løsning.
04.0 KLINISKE OPLYSNINGER
04.1 Terapeutiske indikationer
Filgrastim er indiceret til at reducere varigheden af neutropeni og forekomsten af febril neutropeni hos patienter behandlet med standard cytotoksisk kemoterapi for maligne sygdomme (med undtagelse af kronisk myeloid leukæmi og myelodysplastiske syndromer) og reduktion af varigheden af neutropeni hos patienter, der gennemgår myeloablativ behandling efterfulgt af knoglemarvstransplantation, der anses for at have stor risiko for langvarig alvorlig neutropeni.
Sikkerheden og effekten af filgrastim er ens hos voksne og børn, der gennemgår cytotoksisk kemoterapi.
Filgrastim er indiceret til mobilisering af perifere blod stamceller (PBPC'er).
Hos patienter, børn eller voksne med alvorlig medfødt, cyklisk eller idiopatisk neutropeni, med et absolut neutrofiltal (ANC) ≤ 0,5 x 109 / L, og en historie med alvorlige eller tilbagevendende infektioner, er langvarig administration af filgrastim indiceret til stigende neutrofil tæller og reducerer forekomsten og varigheden af infektionsrelaterede hændelser.
Filgrastim er indiceret til behandling af vedvarende neutropeni (ANC mindre end eller lig med 1,0 x 109 / l) hos patienter med fremskreden hiv -infektion for at reducere risikoen for bakterielle infektioner, når andre behandlingsmuligheder er utilstrækkelige.
04.2 Dosering og indgivelsesmåde
Filgrastim -behandling bør kun udføres i forbindelse med et kræftcenter med
erfaring med G-CSF-behandling og hæmatologi, og hvem der har det nødvendige diagnostiske udstyr. Mobiliserings- og afereseprocedurerne skal udføres i samarbejde med et onkologi-hæmatologisk center med acceptabel erfaring på området, og hvor overvågning af hæmatopoietiske stamceller kan udføres korrekt.
Dosering
Standard cytotoksisk kemoterapi
Den anbefalede dosis filgrastim er 0,5 MU (5 mcg) / kg / dag. Den første dosis filgrastim skal gives mindst 24 timer efter cytotoksisk kemoterapi.
Daglig dosering af filgrastim bør fortsætte, indtil den forventede neutrofile nadir er overskredet, og nuetrofiltallet er vendt tilbage til et normalt niveau.Efter standard kemoterapi for solide tumorer, lymfomer og lymfoide leukæmier kan den krævede behandlingsvarighed for at opfylde disse kriterier nå 14 dage Efter behandling med induktion og konsolidering ved akut myeloid leukæmi kan behandlingsvarigheden være betydeligt længere (op til 38 dage) afhængigt af den anvendte type, dosis og mønster af cytotoksisk kemoterapi.
Hos patienter, der gennemgår cytotoksisk kemoterapi, ses typisk en forbigående stigning i neutrofiltal 1-2 dage efter initiering af filgrastim-behandling. Forventet neutrofil nadir er ikke blevet overskredet, og neutrofiltallet er ikke vendt tilbage til et normalt niveau. For tidlig afbrydelse af behandling med filgrastim, før den forventede neutrofile nadir er nået, anbefales ikke.
Patienter, der gennemgår myeloablativ terapi efterfulgt af knoglemarvstransplantation
Den anbefalede startdosis af filgrastim er 1,0 MU (10 mcg) / kg / dag.
Den første dosis Filgrastim skal administreres mindst 24 timer efter cytotoksisk kemoterapi og mindst 24 timer efter infusion af knoglemarv.
Når neutrofil nadir er passeret, skal den daglige dosis Filgrastim titreres baseret på neutrofilresponsen som følger:
Mobilisering af PBPC'er
Til mobilisering af perifere blodprogenitorceller (PBPC'er) hos patienter, der gennemgår myelosuppressiv eller myeloablativ behandling efterfulgt af autolog perifer blodprogenitorcelletransplantation
Den anbefalede dosis filgrastim til PBPC -mobilisering, når den bruges alene, er 1,0 MU (10 mcg) / kg / dag i 5 til 7 dage i træk. Leukaferese planlægning: 1 eller 2 leukaferese på dag 5 og 6. er ofte tilstrækkelige. I andre tilfælde kan yderligere leukaferese være nødvendig. Filgrastim -administration bør fortsættes indtil den sidste leukaferese.
Den anbefalede dosis filgrastim til PBPC -mobilisering efter myelosuppressiv kemoterapi er 0,5 MU (5 mcg) / kg / dag, der skal administreres dagligt fra den første dag efter endt kemoterapi, indtil den forventede neutrofile nadir er overskredet, og neutrofiltallet ikke er vendt tilbage til et normalt niveau. leukaferese bør udføres i den periode, hvor ANC stiger fra 5,0 x 109 / L. Hos patienter, der ikke gennemgår omfattende kemoterapi, er en enkelt leukaferese ofte tilstrækkelig, i andre tilfælde anbefales yderligere leukaferese.
Til mobilisering af perifere blod progenitorceller (PBPC'er) hos raske donorer før allogen perifer blod stamcelletransplantation
Til PBPC -mobilisering hos raske donorer bør filgrastim administreres ved subkutan injektion i en dosis på 10 mcg / kg / dag i 4 til 5 på hinanden følgende dage. Leukaferese bør begynde på dag 5 og fortsætte efter behov gennem dag 6 for at opnå 4 x 106 CD34 + celler / kg kropsvægt af modtageren.
Hos patienter med alvorlig kronisk neutropeni
Medfødt neutropeni: den anbefalede dosis er 1,2 MU (12 mikrogram) / kg / dag som en enkelt dosis eller i opdelte doser.
Idiopatisk eller cyklisk neutropeni: den anbefalede startdosis er 0,5 MU (5 mcg) / kg / dag som en enkelt dosis eller i opdelte doser.
Dosisjusteringer: Filgrastim bør administreres dagligt, indtil neutrofiltallet er nået og kan opretholdes over 1,5 x 109 / l. Når svaret er opnået, bør den laveste effektive dosis for at opretholde dette niveau bestemmes. Langsigtet daglig administration er påkrævet for at opretholde tilstrækkelige neutrofiltal. Efter en til to ugers behandling kan startdosis fordobles eller halveres baseret på patientens respons. Derefter kan dosis justeres individuelt hver 1-2 uger for at opretholde et gennemsnitligt neutrofiltal mellem 1,5 x 109 / L og 10 x 109 / L. Hos patienter med alvorlige infektioner kan det overvejes en hurtigere tidsplan for progressiv dosiseskalering. I kliniske undersøgelser opnåede 97% af respondenterne et fuldstændigt respons ved doser ≤ 24 mcg / kg / dag. Den langsigtede sikkerhed ved filgrastim-administration ved doser over 24 mikrogram / kg / dag hos patienter med alvorlig kronisk neutropeni er ikke påvist.
Hos patienter med hiv -infektion
Tilbageførsel af neutropeni
Den anbefalede startdosis af filgrastim er 0,1 MU (1 mikrogram) / kg / dag administreret dagligt med titrering op til maksimalt 0,4 MU (4 mcg) / kg / dag, indtil den er nået og kan have et normalt nuetrofiltal (ANC> 2,0 x109 / l) opretholdes. I kliniske undersøgelser reagerede> 90% af patienterne på disse doser og opnåede reversering af neutropeni over en median på 2 dage.
Hos et lille antal patienter (
Opretholdelse af et normalt neutrofiltal
Når reversering af neutropeni er opnået, bør den laveste effektive dosis for at opretholde et normalt neutrofiltal bestemmes. En indledende dosisjustering med alternativ dagsdosis på 30 MU (300 mcg) / dag anbefales. Yderligere dosisjusteringer kan være påkrævet afhængigt af patientens ANC for at opretholde neutrofiltal på> 2,0 x 109 / L. I kliniske forsøg var doser på 30 MU (300 mcg) / dag påkrævet. 1 til 7 dage om ugen at opretholde ANC> 2,0 x 109 / L med en median administrationsfrekvens på 3 dage om ugen. Langsigtet administration kan være påkrævet for at opretholde ANC> 2,0 x 109 / L.
Særlige populationer
Ældre patienter
Kun et lille antal ældre patienter blev inkluderet i kliniske forsøg med filgrastim. Der er ikke udført specifikke undersøgelser i denne patientpopulation. Derfor kan der ikke gives specifikke doseringsanbefalinger til disse patienter.
Patienter med nyre- eller leverinsufficiens
Undersøgelser foretaget med filgrastim hos patienter med svært nedsat nyre- eller leverfunktion viser, at dets farmakokinetiske og farmakodynamiske profil ligner den, der ses hos raske forsøgspersoner. I disse tilfælde er dosisjustering ikke nødvendig.
Pædiatriske patienter med alvorlig kronisk neutropeni (SCN) og maligne sygdomme
I kliniske forsøg var 65 procent af patienterne, der blev behandlet for en SCN, under 18 år. I denne aldersgruppe, herunder hovedsagelig patienter med medfødt neutropeni, er der påvist effekt, og der blev ikke observeret forskelle i sikkerhedsprofiler for pædiatriske patienter, der blev behandlet for alvorlig kronisk neutropeni.
Data fra kliniske forsøg med pædiatriske patienter indikerer, at filgrastims sikkerhed og effekt er ens hos voksne og børn, der gennemgår cytotoksisk kemoterapi.
Doseringsanbefalingerne hos pædiatriske patienter er identiske med anbefalingerne, der gælder for voksne, der gennemgår myelosuppressiv cytotoksisk kemoterapi.
Indgivelsesmåde
Standard cytotoksisk kemoterapi
Filgrastim kan administreres ved daglig subkutan injektion eller daglig intravenøs infusion fortyndet i 50 mg / ml (5%) glucoseopløsning til injektion i løbet af 30 minutter (se afsnit 6.6 om fortyndingsinstruktioner). I de fleste tilfælde foretrækkes den subkutane rute. Der er tegn fra en enkeltdosisundersøgelse på, at intravenøs anvendelse kan reducere virkningens varighed. Den kliniske relevans af dette fund for administration af flere doser er uklar. Valget af indgivelsesvej bør baseres på den enkelte patients kliniske tilstand. I randomiserede kliniske forsøg blev der brugt subkutane doser på 230 mcg / m2 / dag (4,0 til 8,4 mcg / kg / dag).
Patienter, der gennemgår myeloablativ terapi efterfulgt af knoglemarvstransplantation
Filgrastim administreres ved intravenøs infusion, der varer 30 minutter eller ved intravenøs infusion eller ved kontinuerlig 24 timers subkutan infusion. Filgrastim skal fortyndes i 20 ml af en 50 mg / ml (5%) glucoseopløsning til infusion (se pkt. 6.6).
Mobilisering af PBPC'er
Til mobilisering af perifere blodprogitorier (PBPC) hos patienter, der gennemgår myelosuppressiv eller myeloablativ behandling efterfulgt af autolog perifer blodprogenitorcelletransplantation, kan den anbefalede dosis filgrastim ved kontinuerlig subkutan infusion administreres over 24 timer eller ved en enkelt daglig subkutan injektion i 5-7 dage i træk. Til infusion skal filgrastim fortyndes i 20 ml af en 50 mg / ml (5%) glucoseopløsning til injektion (se pkt. 6.6).
NCG / HIV -infektion
Subkutan injektion.
For instruktioner om håndtering af lægemidlet før brug, se afsnit 6.6.
04.3 Kontraindikationer
Overfølsomhed over for det aktive stof eller over for et eller flere af hjælpestofferne nævnt i afsnit 6.1.
04.4 Særlige advarsler og passende forholdsregler ved brug
Særlige advarsler
Filgrastim bør ikke bruges til at øge dosis af cytotoksisk kemoterapi ud over standarddosisregimet.
Filgrastim må ikke anvendes til patienter med alvorlig medfødt neutropeni (Kostmans syndrom) med cytogenetiske abnormiteter.
Overfølsomhedsreaktioner, herunder anafylaktiske reaktioner, der forekommer ved initiering eller efter behandling, er blevet rapporteret hos patienter behandlet med filgrastim. Afbryd behandlingen med filgrastim permanent hos patienter med klinisk signifikant overfølsomhed.Filgrastim må ikke administreres til patienter med en overfølsomhed over for filgrastim eller pegfilgrastim.
Som med alle terapeutiske proteiner er der en potentiel risiko for immunogenicitet. Sandsynligheden for at generere antistoffer mod filgrastim er generelt lav. Udvikling af bindende antistoffer forventes med alle biologiske stoffer, men indtil videre har de ikke været forbundet med aktivitet. Neutralisering.
Spredning af maligne celler
GCSF kan fremme spredning af myeloide celler in vitro og lignende effekter kan ses in vitro på nogle ikke-myeloide celler.
Sikkerheden og effekten af administration af Filgrastim til patienter med myelodysplastisk syndrom eller kronisk myeloid leukæmi er ikke påvist.
Filgrastim er ikke indiceret i sådanne situationer. Der skal lægges særlig vægt på differentialdiagnosen mellem blast -transformation ved kronisk myeloid leukæmi og akut myeloid leukæmi.
På grund af begrænsede data om sikkerhed og effekt bør filgrastim administreres med forsigtighed til patienter med sekundær AML.
Sikkerheden og effekten af filgrastim -administration hos patienter i de novo -alder og gunstig cytogenetik [t (8; 21), t (15; 17) og inv] er ikke påvist.
Andre særlige forholdsregler
Overvågning af knogletæthed kan være indiceret hos patienter med underliggende osteoporose, der er i kontinuerlig filgrastimbehandling i mere end 6 måneder.
Sjældne lungebivirkninger (> 0,01% og interstitiel lungebetændelse er blevet rapporteret efter administration af G-CSF. Patienter med en nylig historie med lungeinfiltration eller lungebetændelse kan have øget risiko. Udseende af lungetegn som hoste, feber og dyspnø i forbindelse med radiologiske tegn på lungeinfiltration og forringelse af lungefunktionen kan være indledende tegn på voksen respiratorisk nødsyndrom (ARDS). Filgrastim bør afbrydes og passende behandling påbegyndes.
Kapillær lækagesyndrom er blevet rapporteret efter administration af granulocytkolonistimulerende faktor og er karakteriseret ved hypotension, hypoalbuminæmi, ødem og hæmokoncentration. Patienter, der udvikler symptomer på kapillær lækagesyndrom, bør overvåges nøje og modtage standard symptomatisk behandling, som kan omfatte behovet for intensiv behandling (se pkt. 4.8).
Særlige forholdsregler hos kræftpatienter
Leukocytose
Hvide blodlegemer på 100 x 109 / l eller mere er blevet observeret hos mindre end 5% af patienterne behandlet med filgrastim i doser over 0,3 MU / kg / dag (3 mcg / kg / dag). Ingen bivirkninger, der direkte kan tilskrives denne grad af leukocytose, blev observeret. I betragtning af de potentielle risici forbundet med alvorlig leukocytose bør der imidlertid foretages regelmæssig overvågning af antallet af hvide blodlegemer under behandling med filgrastim. Filgrastim -behandlingen skal straks stoppes, hvis antallet af hvide blodlegemer overstiger 50 x 109 / l efter den forventede nadir. I perioden med filgrastim -indgivelse til PBPC -mobilisering skal behandlingen stoppes eller dosis reduceres, hvis antallet af hvide blodlegemer overstiger 70 x 109 / l.
Risici forbundet med højdosis kemoterapi
Der bør udvises særlig forsigtighed ved behandling af patienter med højdosis kemoterapi, fordi der ikke er påvist en mere gunstig tumorrespons, og fordi administration af højdosis kemoterapi kan øge toksiske virkninger, herunder hjerte-, pulmonal-, neurologiske og dermatologiske virkninger. (se produktresuméet for de anvendte kemoterapeutiske midler).
Behandling med filgrastim alene forhindrer ikke trombocytopeni og anæmi efter myelosuppressiv kemoterapi. Som følge af muligheden for at modtage højere doser kemoterapi (f.eks. Fulde doser i henhold til det foreskrevne doseringsregime) kan patienten udsættes for en øget risiko for trombocytopeni og anæmi. Det anbefales derfor regelmæssig kontrol af trombocyttal og hæmatokrit.Særlig opmærksomhed bør rettes under administrationen, både alene og i kombination, af kemoterapeutiske midler, der vides at forårsage alvorlig trombocytopeni.
Brugen af filgrastim-mobiliserede PBPC'er har vist sig at reducere sværhedsgraden og varigheden af trombocytopeni efter myelosuppressiv eller myeloablativ kemoterapi.
Splenomegali
Tilfælde af splenomegali og miltbrud er ualmindeligt blevet rapporteret efter administration af filgrastim. Nogle tilfælde af miltbrud har været dødelige. Emner, der modtager filgrastim og rapporterer smerter i venstre øvre del af maven og / eller smerter i skulderekstremitet, bør vurderes for miltforstørrelse eller miltbrud.
Andre særlige forholdsregler
Filgrastims virkning hos patienter med signifikant reducerede myeloide progenitorer er ikke undersøgt. For at øge antallet af neutrofiler virker filgrastim primært på neutrofile forstadier. Derfor hos patienter med et lavt antal prækursorer (f.eks. Hos patienter behandlet med strålebehandling eller omfattende kemoterapi eller patienter med tumorinfiltration af knoglemarven), kan responsen fra neutrofiler reduceres.
Tilfælde af transplantat versus værtsygdom, GvHD og død er blevet rapporteret hos patienter behandlet med G-CSF efter allogen knoglemarvstransplantation (se pkt.5.1).
Filgrastims virkning på GvHD -sygdom er ikke blevet defineret.
Øget knoglemarvs hæmatopoietisk aktivitet som reaktion på vækstfaktorterapi har været forbundet med forbigående positive knogledannelser, hvilket bør overvejes ved fortolkning af knoglerapporter.
Særlige forsigtighedsregler hos patienter, der gennemgår mobilisering af stamceller i perifert blod.
Mobilisering
Der er ingen prospektive radiologiske undersøgelser, der sammenligner de to anbefalede mobiliseringsmetoder (filgrastim alene eller i kombination med myelosuppressiv kemoterapi) inden for den samme patientpopulation. Graden af variation mellem individuelle patienter og mellem laboratorietest af CD34 + celler viser vanskeligheden ved at sammenligne forskellige patienter undersøgelser, derfor er det svært at anbefale en optimal metode. Valget af mobiliseringsmetoden bør evalueres i forhold til de enkelte patientbehandlingsmål.
Tidligere eksponering for cytotoksiske midler
Hos patienter, der er omfattende forbehandlet med myelosuppressiv terapi, er det muligt, at PBPC-mobilisering ikke er tilstrækkelig til at opnå det minimum anbefalede antal celler (2,0 x 106 CD34 + celler / kg), eller at accelerationen af trombocytgenopretning er mindre markant.
Nogle cytotoksiske midler viser særlig toksicitet på hæmatopoietiske stamceller og kan modvirke deres mobilisering. Stoffer som melphalan, carmustine (BCNU) og carboplatin kan, hvis de gives i en længere periode før stamcellemobilisering, reducere antallet af opsamlede celler. Imidlertid har administration af melphalan, carboplatin eller BCNU i kombination med filgrastim vist sig at være effektiv til at mobilisere stamceller. Hvis der planlægges en perifer blodprogenitorcelletransplantation, bør stamcellemobilisering planlægges i den indledende fase af patientens påtænkte behandling. Særlig opmærksomhed bør rettes mod antallet af progenitorceller, der mobiliseres hos sådanne patienter, før administration af højdosis kemoterapi. Hvis celleopsamling er utilstrækkelig i henhold til de tidligere angivne evalueringskriterier, bør alternative behandlinger, der ikke kræver brug af stamceller, overvejes.
Evaluering af samlingen af stamceller
I den kvantitative evaluering af stamceller opnået hos patienter behandlet med filgrastim bør der lægges særlig vægt på opregningsmetoden. Resultaterne af CD34 + celletal ved flowcytometri varierer i henhold til den anvendte metode; derfor skal tal, der stammer fra undersøgelser foretaget i andre laboratorier, fortolkes med forsigtighed.
Statistisk analyse af forholdet mellem antallet af geninfunderede CD34 + -celler og graden af blodpladegenopretning efter højdosis kemoterapi indikerer et komplekst, men konstant forhold.
Anbefalingen om at indsamle et minimumsantal på 2,0 x 106 CD34 + celler / kg er baseret på offentliggjort erfaring, hvilket indikerer, at hæmatologisk genopretning således er tilstrækkelig. Beløb større end det angivne minimumstal synes at være relateret til hurtigere genopretning, mindre beløb til langsommere genopretning.
Særlige forsigtighedsregler for raske donorer, der gennemgår mobilisering af stamceller fra perifert blod
PBPC -mobilisering har ingen direkte klinisk fordel hos raske donorer og bør kun overvejes med henblik på allogen stamcelletransplantation.
PBPC -mobilisering bør kun overvejes hos donorer, der opfylder de normale kliniske og laboratoriekvalificeringskriterier for stamcelledonation, idet der lægges særlig vægt på hæmatologiske parametre og tilstedeværelsen af infektionssygdomme.
Sikkerheden og effekten af filgrastim er ikke blevet evalueret hos raske donorer i alderen 60 år.
Forbigående trombocytopeni (blodplader
Hvis der kræves mere end én leukaferese, donorer med blodplader
Leukaferese bør ikke udføres hos donorer på antikoagulerende behandling eller som har kendt ændringer i hæmostase.
Filgrastim -administration bør afbrydes, eller dosis skal reduceres, hvis antallet af hvide blodlegemer når> 70 x109 / l.
Donorer, der modtager G-CSF til PBPC-mobilisering, bør overvåges, indtil hæmatologiske parametre er normaliseret.
Forbigående cytogene ændringer er blevet observeret efter brug af G-CSF hos raske donorer Betydningen af disse ændringer er ukendt.
Langsigtet sikkerhedsopfølgning hos donorer er i gang. Risikoen for at udvikle en ondartet myeloid celleklon kan dog ikke udelukkes. Det anbefales, at aferesecentret udfører systematisk registrering og screening af stamcelledonorer for at sikre langsigtet sikkerhedsovervågning.
Efter administration af G-CSF er generelt asymptomatisk splenomegali og i meget sjældne tilfælde almindeligt observeret brud på milten hos raske donorer (og patienter). Nogle tilfælde af bristet milt har været dødelige. Derfor skal miltens volumen kontrolleres omhyggeligt (f.eks. Ved fysisk undersøgelse, ultralyd). Diagnosen bristet milt bør overvejes hos donorer og / eller patienter med smerter i venstre øvre del af maven eller smerter i skulderbladet.
Efter markedsføring er der rapporteret meget sjældent om lungebivirkninger (hæmoptyse, lungeblødning, lungeinfiltration, dyspnø og hypoksi) hos normale donorer efter brug af andre lægemidler indeholdende filgrastim. I tilfælde af mistanke om lungebivirkninger eller konstateret, seponering af filgrastim -behandling bør overvejes og nødvendig lægehjælp ydes.
Særlige forsigtighedsregler hos modtagere af allogene celler i perifert blod, der mobiliseres med filgrastim
Aktuelle data indikerer, at immunologiske interaktioner mellem allogene PBPC'er og modtageren kan være forbundet med en øget risiko for akut og kronisk transplantat versus værtsygdom (GvHD) sammenlignet med knoglemarvstransplantation.
Særlige forsigtighedsregler hos patienter med alvorlig kronisk neutropeni (SCN)
Komplet blodtælling
Trombocyttal bør overvåges ofte, især i løbet af de første uger af filgrastim -terapi. Intermitterende seponering af behandlingen eller dosisreduktion af filgrastim bør overvejes hos patienter, der udvikler trombocytopeni, dvs. med blodplader
Andre ændringer i blodbilledet kan forekomme, herunder anæmi og forbigående stigninger i myeloide progenitorer, som kræver omhyggelig overvågning af blodtal.
Transformation til leukæmi eller myelodysplastisk syndrom
Der skal lægges særlig vægt på differentialdiagnosen mellem alvorlig kronisk neutropeni og andre hæmatologiske sygdomme, såsom aplastisk anæmi, myelodispalsi og myeloid leukæmi. Et komplet blodtælling med differential- og trombocyttal samt en evaluering af knoglemarvsmorfologi og en karyotype bør foretages, før behandlingen påbegyndes.
Myelodysplastiske syndromer (MDS) eller leukæmi er blevet observeret hos et lille antal (ca. 3%) af patienter med alvorlig kronisk neutropeni behandlet med filgrastim i kliniske forsøg. Dette er kun blevet observeret hos patienter med medfødt neutropeni. MDS og leukæmi er naturlige komplikationer af sygdommen og skal ikke overvejes med sikkerhed i forbindelse med behandling med filgrastim. Abnormiteter, herunder monosomi 7, blev efterfølgende fundet hos ca. 12% af patienterne med normal cytogenetik ved baseline under rutinemæssig gentagen test. Hvis patienter med alvorlig kronisk neutropeni udvikler cytogenetiske abnormiteter, bør risici og fordele ved fortsat behandling med filgrastim overvejes; Filgrastim -administration bør afbrydes, hvis MDS eller leukæmi udvikler sig. Det er i øjeblikket ukendt, om langvarig behandling af patienter med alvorlig kronisk neutropeni kan disponere patienter for cytogenetiske abnormiteter, MDS eller leukæmisk transformation. Hos disse patienter anbefales morfologiske og cytogenetiske analyser af knoglemarven med jævne mellemrum (cirka hver 12. måned).
Splenomegali
Tilfælde af splenomegali og miltbrud er ualmindeligt blevet rapporteret efter administration af filgrastim. Nogle tilfælde af miltbrud har været dødelige. Emner, der modtager filgrastim og rapporterer smerter i venstre øvre del af maven og / eller smerter i skulderekstremitet, bør vurderes for miltforstørrelse eller miltbrud.
Andre særlige forholdsregler
Årsager til forbigående neutropeni, såsom virusinfektioner, skal udelukkes.
Splenomegali er en direkte effekt af filgrastim -behandling. Mærkbar splenomegali blev observeret hos 31% af patienterne i kliniske undersøgelser. Volumenforøgelser målt radiologisk blev set tidligt under filgrastim -terapi og viste en tendens til at stabilisere sig. Dosisreduktioner blev observeret for at bremse eller standse udviklingen af splenomegali, og en splenektomi var påkrævet hos 3% af patienterne. Miltens volumen bør kontrolleres regelmæssigt. Palpation i maven er tilstrækkelig til at detektere unormale volumenstigninger.
Hæmaturi / proteinuri forekom hos et lille antal patienter. Urinalyse bør udføres med jævne mellemrum for at opdage sådanne hændelser.
Sikkerhed og effekt hos nyfødte og hos patienter med autoimmun neutropeni er ikke påvist.
Særlige forholdsregler hos HIV -inficerede patienter
Komplet blodtælling
Absolut neutrofiltal (ANC) bør overvåges ofte, især i løbet af de første uger af filgrastim -behandlingen. Nogle patienter kan reagere meget hurtigt og med en markant stigning i neutrofiltal ved startdosis af filgrastim. Det anbefales, at ANC bestemmes dagligt i løbet af de første 2-3 dage efter filgrastim-administration. Derefter anbefales det, at ANC bestemmes mindst to gange om ugen i løbet af de første 2 uger og en gang om ugen eller hver anden uge derefter. under vedligeholdelsesbehandling. Ved intermitterende administration af 30 MU (300 mcg) / dag af filgrastim kan der forekomme store udsving over tid i ANC. For at bestemme minimums- eller nadirværdien af en patients ANC anbefales det, at der tages blodprøver. Beregnet til ANC -bestemmelse umiddelbart før den påtænkte administration af filgrastim.
Risici forbundet med høje doser af myelosuppressive lægemidler
Behandling med filgrastim alene forhindrer ikke trombocytopeni og anæmi efter myelosuppressiv behandling. Da højere doser eller flere af disse lægemidler kan administreres ved brug af filgrastim, kan patienten have en øget risiko for trombocytopeni eller anæmi. Regelmæssig monitorering af hæmatokrit anbefales (se ovenfor).
Infektioner og maligniteter forårsager myelosuppression
Et neutropeni kan skyldes knoglemarvsinfiltration fra opportunistiske infektioner, som f.eks Mycobacterium avium komplekse eller til maligne neoplasmer, såsom lymfomer. Hos patienter med kendte knoglemarvsinfiltrerende infektioner eller maligniteter bør der overvejes passende behandling af den underliggende sygdom ud over administration af filgrastim til behandling af neutropeni. Filgrastims virkninger på neutropeni på grund af knoglemarvsinfiltrerende infektioner eller maligniteter er ikke endeligt påvist.
Splenomegali
Tilfælde af splenomegali og miltbrud er ualmindeligt blevet rapporteret efter administration af filgrastim. Nogle tilfælde af miltbrud har været dødelige. Emner, der modtager filgrastim og rapporterer smerter i venstre øvre del af maven og / eller smerter i skulderekstremitet, bør vurderes for miltforstørrelse eller miltbrud.
Særlige forholdsregler ved seglcelle- eller seglcelleanæmi
Seglcellekriser, i nogle tilfælde dødelige, er blevet rapporteret hos patienter med seglcelle- eller seglcellesygdom behandlet med filgrastim. Hos patienter med seglcelleegenskab eller seglcelleanæmi bør læger udvise forsigtighed, når de vurderer brugen af filgrastim, som kun bør anvendes efter omhyggelig overvejelse af de potentielle fordele og risici.
Hjælpestoffer
Nivestim indeholder sorbitol. Patienter med sjælden arvelig fructoseintolerance bør ikke bruge denne medicin. Den indeholder også mindre end 1 mmol natrium (23 mg) pr. Dosis, dvs. i det væsentlige "natriumfrit".
04.5 Interaktioner med andre lægemidler og andre former for interaktion
Sikkerheden og effekten af filgrastim administreret på samme dag som myelosuppressiv cytotoksisk kemoterapi er ikke endeligt påvist. Da hurtigt delende myeloide celler er følsomme for myelosuppressiv cytotoksisk kemoterapi, anbefales det ikke at bruge filgrastim i perioden. Mellem 24 timer før og 24 timer efter kemoterapi. Foreløbige data indhentet hos et lille antal patienter behandlet i fællesskab med filgrastim og 5-Fluorouracil indikerer, at neutropeni kan forværres.
De mulige interaktioner med andre hæmatopoietiske vækstfaktorer og cytokiner er endnu ikke undersøgt i kliniske forsøg.
Da lithium fremmer frigivelsen af neutrofiler, vil det sandsynligvis forstærke effekten af filgrastim. Selvom denne interaktion ikke er formelt undersøgt, er der ingen tegn på, at den er skadelig.
04.6 Graviditet og amning
Graviditet
Der er ingen eller begrænsede data om brugen af filgrastim til gravide.
Dyrestudier har vist reproduktionstoksicitet. En "øget forekomst af aborter efter eksponering" for høje multipler af kliniske doser blev observeret hos kaniner
og i nærvær af maternel toksicitet (se pkt. 5.3). Tilfælde er blevet beskrevet i litteraturen, hvor der er påvist placenta -spredning af filgrastim hos gravide kvinder. Filgrastim anbefales ikke under graviditet.
Fodringstid
Det vides ikke, om filgrastim udskilles i modermælk; derfor anbefales filgrastim ikke til kvinder, der ammer. Der skal træffes en beslutning om, hvorvidt amning skal afbrydes eller afbrydes / afstå fra behandling med filgrastim under hensyntagen til fordelen ved amning for barnet og fordelen ved behandling for kvinden.
Fertilitet
Filgrastim havde ingen effekt på reproduktion eller fertilitet hos han- eller hunrotter (se pkt. 5.3).
04.7 Virkninger på evnen til at føre motorkøretøj og betjene maskiner
Filgrastim har beskedne virkninger på evnen til at føre motorkøretøj og betjene maskiner.Hvis patienten føler sig træt, skal man være forsigtig, når man kører bil eller betjener maskiner.
04.8 Bivirkninger
Resumé af sikkerhedsprofilen
I kliniske forsøg blev 183 kræftpatienter og 96 raske frivillige udsat for Nivestim.
Sikkerhedsprofilen for filgrastim observeret i disse kliniske undersøgelser var i overensstemmelse med den, der blev rapporteret for det referenceprodukt, der blev brugt i disse undersøgelser.
I kliniske undersøgelser af kræftpatienter var bivirkningen, der kan tilskrives filgrastim ved den hyppigst anbefalede dosis, muskuloskeletale smerter, mild eller moderat hos 10% og alvorlig hos 3% af patienterne.
Graft versus værtreaktionssygdom (GvHD) er også blevet rapporteret (se nedenfor).
Ved mobilisering af perifere cirkulerende stamceller (PBPC'er) hos raske donorer var den mest almindelige rapporterede bivirkning muskuloskeletale smerter. Leukocytose blev observeret hos donorer, og trombocytopeni efter filgrastim, og leukaferese blev også observeret hos donorer. Splenomegali og miltbrud er også blevet rapporteret. Nogle tilfælde af miltbrud har været dødelige.
Hos patienter med alvorlig kronisk neutropeni (SCN) var de hyppigste bivirkninger, der kan tilskrives filgrastim, knoglesmerter, generelle muskuloskeletale smerter og splenomegali.
Kapillær lækagesyndrom, som kan være livstruende, hvis behandlingen forsinkes, er blevet rapporteret ualmindeligt (≥1 / 1000 til
I kliniske forsøg med HIV -patienter var muskuloskeletale smerter, milde til moderate knoglesmerter og myalgi de eneste bivirkninger, der blev anset for at være unikt relateret til filgrastim -administration. Forekomsten af disse reaktioner lignede den, der blev rapporteret hos kræftpatienter.
Tabel over bivirkninger
Bivirkningerne anført nedenfor og deres hyppighed blev observeret efter behandling med filgrastim ifølge offentliggjorte data.
Bivirkningernes hyppighed er defineret i henhold til følgende konventioner:
Meget almindelig: ≥1 / 10
Almindelig: ≥1 / 100 y
Ikke almindelig: ≥1 / 1.000 y
Sjælden: ≥1 / 10.000 y
Meget sjælden:
Ikke kendt: kan ikke estimeres ud fra de tilgængelige data
Inden for hver frekvensgruppe præsenteres bivirkninger i faldende sværhedsgrad.
Hos kræftpatienter
Sunde donorer, der gennemgår mobilisering af perifere blodprogenitorceller
Hos patienter med alvorlig kronisk neutropeni (SCN)
Hos patienter med hiv
Beskrivelse af udvalgte bivirkninger
GvHD og dødsfald er blevet rapporteret hos patienter, der får G-CSF efter allogen knoglemarvstransplantation (se pkt.5.1).
Tilfælde af kapillær lækagesyndrom er blevet rapporteret efter markedsføring med brug af granulocytkolonistimulerende faktorer, som generelt er forekommet hos patienter med fremskreden malign sygdom, sepsis, der tog flere kemoterapilægemidler eller gennemgik aferes (se pkt. 4.4).
Hos kræftpatienter
Muskuloskeletale smerter kontrolleres normalt med standard analgetika. Mindre hyppige bivirkninger inkluderer urinabnormiteter med overvejende mild eller moderat dysuri.
I randomiserede, placebokontrollerede forsøg øgede filgrastim ikke forekomsten af bivirkninger forbundet med cytotoksisk kemoterapi Bivirkninger observeret med samme hyppighed hos patienter behandlet med filgrastim kemoterapi og placebo / kemoterapi var kvalme, opkastning, alopeci, diarré, træthed, anoreksi , hovedpine, hosteudslæt, brystsmerter, generel svaghed, ondt i halsen, forstoppelse og uspecificerede smerter.
Reversible, dosisafhængige og normalt milde til moderate stigninger i lactatdehydrogenase, alkalisk phosphatase, urinsyre er blevet rapporteret hos henholdsvis 50%, 35%, 25%og 10%af patienterne behandlet med filgrastim i anbefalede doser. Serum og gamma -glutamyltranspeptidase.
Der er også lejlighedsvis blevet rapporteret forbigående stigninger i blodtrykket, der ikke kræver nogen klinisk behandling.
Karsygdomme, herunder veno-okklusiv sygdom og ændringer i væskemængde, er lejlighedsvis blevet rapporteret hos patienter behandlet med høje doser kemoterapi efterfulgt af autolog knoglemarvstransplantation. Et årsagssammenhæng med filgrastim er ikke påvist.
Sjældne bivirkninger af kutan vaskulitis er blevet rapporteret hos patienter behandlet med filgrastim. Vaskulitis mekanisme hos patienter behandlet med filgrastim kendes ikke.
Lejlighedsvise tilfælde af Sweet's syndrom (akut febril dermatose) er blevet beskrevet. Da en betydelig procentdel af disse patienter blev diagnosticeret med leukæmi, en tilstand, der vides at være forbundet med Sweet's syndrom, er der imidlertid ikke påvist et årsagssammenhæng med filgrastim.
I enkelte tilfælde er der rapporteret om en "forværring af" leddegigt.
Der har været sjældne rapporter om lungebivirkninger, herunder interstitiel lungebetændelse, lungeødem og lungeinfiltrationer med respirationssvigt eller voksen respiratorisk nødsyndrom (ARDS), som kan være dødelig (se pkt. 4.4).
Allergiske reaktioner: Allergiske reaktioner, herunder anafylaksi, udslæt, urticaria, angioødem, dyspnø og hypotension, der forekommer hos patienter på første eller efterfølgende filgrastim-behandling, er blevet rapporteret. Samlet set var rapporter hyppigere efter administration. I nogle tilfælde har symptomer gentages ved genoptagelse af behandlingen, hvilket tyder på en årsagssammenhæng Hos patienter, der oplever en alvorlig allergisk reaktion på filgrastim, bør behandlingen afbrydes permanent.
Der er rapporteret om isolerede tilfælde af seglcellekriser hos patienter med seglcelle- eller seglcelleanæmi (se pkt. 4.4). Ud fra kliniske data anslås hyppigheden som usædvanlig.
Pseudogout er blevet rapporteret hos kræftpatienter behandlet med filgrastim.
Sunde donorer, der gennemgår perifer blodprogenitorcellemobilisering
Leukocytose (antal hvide blodlegemer (WBC)> 50 x 109 / l) blev rapporteret hos 41% af donorer og trombocytopeni (blodplader
Forbigående og milde stigninger i alkalisk fosfatase, LDH, SGOT og urinsyre er blevet rapporteret hos raske donorer behandlet med filgrastim, som ikke havde kliniske følger.
En "forværring af artritiske symptomer er blevet rapporteret meget sjældent.
Alvorlige allergiske reaktioner er blevet rapporteret meget sjældent.
Hovedpine, der menes at være udløst af filgrastim, er blevet rapporteret i undersøgelser med raske PBPC -donorer.
Asymptomatiske tilfælde af splenomegali og meget sjældne tilfælde af miltbrud er almindeligt blevet rapporteret hos raske donorer og hos patienter efter administration af granulocytkolonistimulerende faktorer (G-CSF'er) (se pkt.4.4).
Lungebivirkninger hos raske donorer (hæmoptyse, lungeblødning, lungeinfiltrater, dyspnø og hypoxi) er meget sjældent rapporteret efter markedsføring af andre filgrastimlægemidler (se pkt. 4.4).
Hos patienter med alvorlig kronisk neutropeni (SCN)
Bivirkninger relateret til filgrastimbehandling er blevet rapporteret hos SCN -patienter, og for nogle af dem har deres hyppighed tendens til at falde over tid.
Bivirkninger omfatter splenomegali, der kan være progressiv i et mindretal af tilfældene og trombocytopeni. Hovedpine og diarré er normalt blevet rapporteret hos mindre end 10% af patienterne kort tid efter initiering af filgrastim -behandling. Anæmi og epistaxis er også blevet rapporteret.
Forbigående stigninger i serum uden kliniske symptomer er blevet rapporteret for urinsyre, lactatdehydrogenase og alkalisk phosphatase.Der er også rapporteret om forbigående og moderate fald i ikke-fastende blodglukose.
Blandt de bivirkninger, der sandsynligvis vedrørte filgrastim -behandling, der normalt forekom ved artralgi, alopeci, osteoporose og udslæt.
Kutan vaskulitis blev rapporteret hos 2% af SCN -patienterne efter længere tids brug. Der har kun været få tilfælde af proteinuri / hæmaturi.
Hos patienter med hiv
Splenomegali blev rapporteret at være sekundær til filgrastimbehandling ved hypersplenisme, og ingen patient gennemgik miltomi. Forholdet til filgrastim-behandling er ukendt, da splenomegali er almindelig hos HIV-inficerede patienter og er til stede i varierende grad hos de fleste AIDS-patienter.
Pædiatrisk population
Data fra kliniske forsøg med pædiatriske patienter indikerer, at filgrastims sikkerhed og effekt er ens hos både voksne og børn, der får cytotoksisk kemoterapi, hvilket tyder på, at der ikke er aldersrelaterede forskelle i filgrastims farmakokinetik. Den eneste bivirkning, der konsekvent blev rapporteret, var muskuloskeletale smerter, som ikke adskiller sig fra oplevelsen hos den voksne befolkning.
Der er utilstrækkelige data til yderligere at evaluere brugen af filgrastim til pædiatriske forsøgspersoner.
Andre særlige populationer
Geriatrisk brug
Generelt blev der ikke observeret forskelle i sikkerhed eller effekt mellem forsøgspersoner over 65 år og yngre voksne (> 18 år), der modtog cytotoksisk kemoterapi og klinisk erfaring, identificerede ingen forskelle i respons mellem ældre og yngre voksne patienter. Der er utilstrækkelige data til at evaluere brugen af filgrastim hos geriatriske personer til de andre godkendte indikationer af filgrastim.
Pædiatriske patienter med alvorlig kronisk neutropeni (SNG)
Der er rapporteret tilfælde af nedsat knogletæthed og osteoporose hos pædiatriske patienter med alvorlig kronisk neutropeni, der modtog kronisk behandling med filgrastim. Hyppigheden anslås som "almindelig" ud fra data fra kliniske forsøg.
Indberetning af formodede bivirkninger
Rapportering af formodede bivirkninger, der opstår efter godkendelse af lægemidlet, er vigtig, da det giver mulighed for kontinuerlig overvågning af lægemidlets fordel / risiko -balance.Professionelle sundhedspersonale anmodes om at rapportere alle formodede bivirkninger via det nationale rapporteringssystem. "Bilag V .
04.9 Overdosering
Virkningerne af filgrastim -overdosis er ikke påvist.
Afbrydelse af filgrastim-behandlingen resulterer normalt i et fald på 50% i cirkulerende neutrofiler inden for 1-2 dage og vender tilbage til det normale inden for 1-7 dage.
05.0 FARMAKOLOGISKE EGENSKABER
05.1 Farmakodynamiske egenskaber
Farmakoterapeutisk gruppe: immunstimulerende, kolonistimulerende faktorer.
ATC -kode: L03AA02.
Nivestim er et biosimilært lægemiddel. Yderligere oplysninger er tilgængelige på webstedet for Det Europæiske Lægemiddelagentur www.ema.europa.eu
Human G-CSF er et glycoprotein, der regulerer produktionen og frigivelsen af funktionelle neutrofiler fra knoglemarven. Nivestim, der indeholder r-metHuG-CSF (filgrastim), fremkalder en markant stigning i neutrofiltal i perifert blod og en mindre markant stigning i monocytter inden for 24 timer. Hos nogle patienter med alvorlig kronisk neutropeni kan filgrastim forårsage endda en lille stigning i antallet af cirkulerende eosinofiler og basofiler fra baseline; nogle af disse patienter kan have eosinofili eller basofili allerede før behandling. Ved anbefalede doser er stigningen i antallet af neutrofiler dosisafhængig.Som det fremgår af de udførte analyser, viser neutrofiler produceret som reaktion på filgrastim normale eller øgede kemotaktiske og fagocytiske egenskaber. Efter afslutning af filgrastim-behandling falder antallet af cirkulerende neutrofiler med ca. 50% inden for 1-2 dage og når normale niveauer inden for 1-7 dage.
Anvendelsen af filgrastim til patienter, der gennemgår cytotoksisk kemoterapi, reducerer signifikant forekomsten, sværhedsgraden og varigheden af nuetropeni og febril neutropeni. Behandling med filgrastim reducerer signifikant varigheden af febril neutropeni, brug af antibiotika og hospitalsindlæggelse efter induktionskemoterapi ved akut myeloid leukæmi eller myeloablativ behandling efterfulgt af knoglemarvstransplantation. I begge tilfælde var forekomsten af feber og dokumenterede infektioner ikke reduceret. Febervarighed blev ikke reduceret hos patienter, der gennemgik myeloablativ behandling efterfulgt af knoglemarvstransplantation.
Anvendelsen af filgrastim, enten alene eller efter kemoterapi, mobiliserer hæmatopoietiske stamceller i perifert blod Disse autologe perifere blodprogenitorceller (PBPC'er) kan opsamles og geninfunderes efter højdosis cytotoksisk kemoterapi, som et alternativ til eller ud over transplantation af knoglemarv. PBPC -infusion fremskynder hæmatopoietisk genopretning og reducerer dermed varigheden af risikoen for blødningskomplikationer og behovet for trombocyttransfusioner.
Modtagere af perifere allogene blodprogenitorceller mobiliseret med filgrastim, sammenlignet med allogene knoglemarvstransplantationsmodtagere, rapporterede en signifikant hurtig hæmatologisk genopretning med deraf følgende signifikant genopretning af tid uden forsyning af trombocytter.
En retrospektiv europæisk undersøgelse, hvor brugen af G-CSF efter allogen knoglemarvstransplantation hos patienter med akut leukæmi blev analyseret, indikerede en øget risiko for GvHD og dødelighed efter administration af G-CSF (TRM I en anden international retrospektiv undersøgelse, udført med patienter med akutte og kroniske myeloide leukæmier, blev der ikke observeret nogen effekt på risikoen for GvHD, TRM og dødelighed.I en metaanalyse af allogene transplantationsundersøgelser, herunder resultaterne af ni prospektive randomiserede undersøgelser, 8 retrospektive undersøgelser og 1 case-control undersøgelse, blev der ikke observeret nogen effekt på risikoen for akut GvHD, kronisk GvHD eller tidlig behandlingsrelateret dødelighed.
a Analysen omfatter undersøgelser vedrørende knoglemarvstransplantation i den pågældende periode; GM-CSF blev brugt i nogle undersøgelser
b Analysen omfatter patienter, der gennemgik knoglemarvstransplantation i den pågældende periode
Anvendelse af filgrastim til PBPC -mobilisering hos raske donorer før allogen PBPC -transplantation resulterer i genopretning af 4 x 106 CD34 + celler / kg pr. Modtagerlegemsvægt hos de fleste donorer efter to leukaferer. Normale donorer får en dosis på 10 mcg / kg / dag subkutant i 4-5 på hinanden følgende dage.
Anvendelse af filgrastim til patienter, voksne og børn med svær kronisk neutropeni (alvorlig medfødt, cyklisk og idiopatisk neutropeni) medfører en markant stigning i det neutrale neutrofiltal i perifert blod og et fald i infektionsepisoder og relaterede hændelser.
Brugen af filgrastim til HIV-inficerede patienter opretholder neutrofiltal på normale niveauer og tillader således, at de antivirale og / eller myelosuppressive lægemidler administreres efter hensigten. Der er ingen tegn på, at HIV-inficerede patienter og behandlet med filgrastim viser øget HIV-replikation.
Som det er blevet observeret med andre hæmatopoietiske vækstfaktorer, viser G-CSF også in vitro en stimulerende virkning på humane endotelceller.
Nivestims effekt og sikkerhed er blevet undersøgt i randomiserede, kontrollerede fase III -undersøgelser af brystkræft Der blev ikke fundet relevante forskelle mellem Nivestim og referenceproduktet med hensyn til varigheden af alvorlig neutropeni og forekomsten af febril neutropeni.
05.2 Farmakokinetiske egenskaber
Et randomiseret, ukrypteret, enkeltdosis, komparatorstyret forsøg, i to eksemplarer crossover udført på 46 raske frivillige viste, at Nivestims farmakokinetiske profil var sammenlignelig med referenceproduktets efter subkutan og intravenøs behandling.
I en anden randomiseret, dobbeltblind, multiple-dosis, komparator-kontrolleret, dobbelt-crossover-undersøgelse på 50 raske frivillige viste, at Nivestims farmakokinetiske profil var sammenlignelig med referenceproduktets efter subkutan behandling.
Clearance af filgrastim har vist sig at følge førsteordens farmakokinetik efter både subkutan og intravenøs behandling. Serumelimineringshalveringstiden for filgrastim er cirka 3,5 timer med en eliminationshastighed på cirka 0,6 ml / min / kg.Kontinuerlig infusion af filgrastim over en periode på 28 dage hos patienter med restitutionsfase fra autolog knoglemarvstransplantation viste ikke enhver ophobning af lægemidlet med en sammenlignelig halveringstid. Derfor er der en positiv lineær sammenhæng mellem dosis og serumkoncentration af filgrastim, uanset om det administreres intravenøst eller subkutant. Efter subkutan administration af de anbefalede doser blev serumkoncentrationerne holdt over 10 ng / ml i 8 - 16 timer. Distributionsvolumen i blodet er cirka 150 ml / kg.
05.3 Prækliniske sikkerhedsdata
Filgrastim er blevet undersøgt i toksicitetsstudier med gentagen dosis op til 1 års varighed, hvilket afslørede ændringer, der kan tilskrives forventede farmakologiske virkninger, herunder øgede leukocytter, myeloid knoglemarvshyperplasi, ekstramedullær granulocytopoiesis og miltforstørrelse.
Disse ændringer er alle reversible efter behandlingens ophør.
Filgrastims virkninger på prænatal udvikling blev undersøgt hos rotter og kaniner. Intravenøs administration (80 mikrogram / kg / dag) af filgrastim til kaniner i organogeneseperioden viste toksicitet hos moderen og en stigning i spontane aborter, tab efter implantation og fald i gennemsnitlig levende kuldstørrelse og fostervægt.
Baseret på data rapporteret for et andet filgrastim-produkt, blev lignende resultater observeret ud over "øgede fostermisdannelser ved en dosis på 100 mcg / kg / dag, en maternel toksicitetsdosis svarende til en" systemisk eksponering på ca. 50-90 gange den eksponering, der blev observeret i patienter behandlet med den kliniske dosis på 5 mcg / kg / dag. Det niveau, hvor der ikke blev observeret nogen negativ effekt på embryo-fostertoksicitet i denne undersøgelse, var 10 mcg / kg / dag, hvilket svarede til en systemisk eksponering cirka 3-5 gange eksponeringen observeret hos patienter behandlet med den kliniske dosis.
Hos drægtige rotter blev der ikke observeret moder- eller fostertoksicitet ved doser over 575 mcg / kg / dag. Administration af filgrastim til afkom af rotter i peri-natal- og diegivningsperioderne viste en forsinkelse i ekstern differentiering og væksthæmning (≥ 20 mcg / kg / dag) og en let reduceret overlevelsesrate (100 mcg / kg / dag).) .
Der blev ikke observeret effekter på fertiliteten hos han- eller hunrotter for filgrastim.
06.0 LÆGEMIDDELOPLYSNINGER
06.1 Hjælpestoffer
Eddikesyre, glacial
Natriumhydroxid
Sorbitol (E420)
Polysorbat 80
Vand til injektionsvæsker
06.2 Uforenelighed
Nivestim må ikke fortyndes med natriumchloridopløsninger.
Fortyndet filgrastim kan absorberes af glas og plast, medmindre det fortyndes i en 50 mg / ml (5%) glucoseopløsning til infusion (se pkt. 6.6).
Dette lægemiddel må ikke blandes med andre lægemidler, undtagen dem, der er anført i afsnit 6.6.
06.3 Gyldighedsperiode
Forfyldt sprøjte
30 måneder.
Efter fortynding
Kemisk og fysisk stabilitet i den fortyndede infusionsvæske, opløsning er blevet påvist i 24 timer ved 2 ° C til 8 ° C. Fra et mikrobiologisk synspunkt bør produktet bruges med det samme. Hvis lægemidlet ikke bruges med det samme, er brugeren ansvarlig for opbevaringens varighed og betingelser før brug; lægemidlet kan opbevares i op til 24 timer ved en temperatur mellem 2 ° C og 8 ° C, medmindre fortynding finder sted under kontrollerede og validerede aseptiske forhold.
06.4 Særlige opbevaringsforhold
Opbevares og transporteres køligt (2 ° C - 8 ° C).
Må ikke fryses.
Opbevar de fyldte sprøjter i den ydre karton for at beskytte dem mod lys.
Utilsigtet udsættelse for frysetemperaturer i op til 24 timer har ingen negativ indvirkning på Nivestims stabilitet.Den frosne fyldte injektionssprøjte kan optøs og returneres til køleskabet til senere brug. Hvis eksponeringen for lave temperaturer er større end 24 timer, eller hvis den er frosset mere end én gang, må Nivestim IKKE bruges mere.
Inden for dets holdbarhed og til ambulant brug kan patienten fjerne produktet fra køleskabet og opbevare det ved stuetemperatur (ikke over 25 ° C) i en gang og op til 7 dage. Produktet må ikke længere anbringes i køleskabet og skal kasseres.
Opbevaringsbetingelser for lægemidlet efter fortynding, se afsnit 6.3.
06.5 Den umiddelbare emballages art og emballagens indhold
Forfyldt sprøjte (glas af type I) med injektionsnål (rustfrit stål) med kanylebeskyttelse, der indeholder 0,2 ml injektions- / infusionsvæske, opløsning.
Pakningsstørrelse på 1, 5 eller 10 fyldte sprøjter.
Ikke alle pakningsstørrelser er nødvendigvis markedsført.
06.6 Brugsanvisning og håndtering
Om nødvendigt kan Nivestim fortyndes i 50 mg / ml (5%) glucoseopløsning til injektion.
Fortynding til en slutkoncentration under 0,2 MU (2 mcg) pr. Ml anbefales aldrig.
Opløsningen bør inspiceres visuelt før brug. Der bør kun bruges klare, partikelfrie opløsninger.
Hos patienter behandlet med filgrastim fortyndet til koncentrationer under 1,5 MU (15 mcg) pr. Ml, skal humant serumalbumin (HSA) tilsættes i en slutkoncentration på 2 mg / ml.
Eksempel: I et sidste volumen, der skal injiceres på 20 ml, bør totale doser filgrastim under 30 MU (300 mcg) administreres ved tilsætning af 0,2 ml 20% human albuminopløsning.
Filgrastim fortyndet i glukoseopløsning ved 50 mg / ml (5%) er kompatibelt med glas og mange plastmaterialer, herunder PVC, polyolefin (en copolymer af polypropylen og polyethylen) og polypropylen.
Nivestim indeholder ikke konserveringsmidler. På grund af risikoen for bakteriel kontaminering er Nivestim sprøjter kun til engangsbrug. Ubrugt lægemiddel og affald fra dette lægemiddel skal bortskaffes i overensstemmelse med lokale bestemmelser.
07.0 INDEHAVER AF MARKEDSFØRINGSTILLADELSE
Hospira UK Limited
Queensway
Royal Leamington Spa
Warwickshire CV31 3RW
Storbritannien
08.0 MARKEDSFØRINGSTILLADELSESNUMMER
EU/1/10/631/001
040158014
EU/1/10/631/002
040158026
EU/1/10/631/003
040158038
09.0 DATO FOR FØRSTE TILLADELSE ELLER FORNYELSE AF TILLADELSEN
Dato for første godkendelse: 8. juni 2010
10.0 DATO FOR REVISION AF TEKSTEN
D.CCE maj 2015