Aktive ingredienser: Tranexamsyre
UGUROL 500 mg / 5 ml injektionsvæske, opløsning til intravenøs anvendelse, til oral og lokal brug
Ugurol indlægssedler er tilgængelige til pakninger:- UGUROL 500 mg / 5 ml injektionsvæske, opløsning til intravenøs anvendelse, til oral og lokal brug
- UGUROL 250 mg tabletter
Hvorfor bruges Ugurol? Hvad er det for?
UGUROL indeholder tranexaminsyre, som tilhører en gruppe lægemidler kaldet antihemoragiske midler, antifibrinolytika, aminosyrer.
UGUROL bruges til voksne og børn fra et års alder til at forebygge og behandle blødning på grund af en proces, der hæmmer blodpropper kaldet fibrinolyse.
De specifikke indikationer er:
- tung menstruationscyklus;
- gastrointestinal blødning;
- blødningsforstyrrelser i urinvejene, efter prostata- eller urinvejskirurgi;
- hjerte, mave eller gynækologisk operation
- blødning efter behandling med anden medicin for at opløse blodpropper.
Kontraindikationer Når Ugurol ikke bør bruges
Tag ikke UGUROL:
- hvis du er allergisk over for tranexaminsyre eller et af de øvrige indholdsstoffer i denne medicin
- hvis du har en sygdom, der fører til blodpropper
- hvis du har en tilstand kaldet 'forbrugskoagulopati', hvor blod begynder at størkne i forskellige dele af kroppen
- hvis du har nyreproblemer
- hvis du tidligere har haft anfald.
På grund af risikoen for cerebralt ødem og anfald anbefales intratekal og intraventrikulær injektion og intracerebral anvendelse.
Hvis du mener, at noget af dette gælder for dig, eller hvis du har andre spørgsmål, skal du tale med din læge, før du tager UGUROL.
Forholdsregler ved brug Det, du skal vide, før du tager Ugurol
Fortæl din læge, hvis noget af følgende gælder for dig for at hjælpe dem med at beslutte, om UGUROL er det rigtige for dig:
- hvis du har bemærket blod i urinen, kan det skyldes en obstruktion i urinvejene;
- hvis du er i risiko for blodpropper
- hvis du har overdreven blodpropper eller blødninger i hele din krop (spredt intravaskulær koagulation), er UGUROL muligvis ikke egnet til dig, medmindre du har akut alvorlig blødning og blodprøver har vist, at processen, der hæmmer blodpropper, kaldet fibrinolyse, aktiveres;
- hvis du har haft anfald, bør UGUROL ikke gives. Din læge bør bruge den lavest mulige dosis for at undgå anfald på grund af behandling med UGUROL;
- hvis du er i langvarig behandling med UGUROL, skal du være opmærksom på mulige forstyrrelser i farvesynet, og om nødvendigt skal behandlingen stoppes. I tilfælde af langvarig brug af UGUROL injektionsvæske, anbefales regelmæssige oftalmologiske undersøgelser (øjenundersøgelser, herunder synsskarphed, farvesyn, fundus, synsfelt osv.). I tilfælde af patologiske oftalmologiske ændringer, især med nethindesygdomme, skal din læge, efter at have konsulteret en specialist, beslutte behovet for langvarig brug af UGUROL injektionsvæske, opløsning i dit tilfælde.
Interaktioner Hvilke lægemidler eller fødevarer kan ændre virkningen af Ugurol
Fortæl det til din læge eller apotek, hvis du tager eller for nylig har taget anden medicin, herunder receptpligtig, vitaminer, mineraler, naturlægemidler eller kosttilskud.
Du skal især fortælle det til din læge, hvis du tager:
- anden medicin, der hjælper blodpropper kaldet antifibrinolytika;
- medicin, der forhindrer blodpropper kaldet trombolytika;
- orale præventionsmidler.
Advarsler Det er vigtigt at vide, at:
Graviditet og amning
Spørg din læge til råds, hvis du er gravid eller ammer, før du tager UGUROL.
Tranexaminsyre udskilles i modermælk, derfor anbefales brug af UGUROL ikke under amning.
Kørsel og brug af maskiner
Der er ikke udført undersøgelser af evnen til at føre motorkøretøj eller betjene maskiner.
Dosis, metode og administrationstidspunkt Sådan bruges Ugurol: Dosering
Tag denne medicin nøjagtigt efter lægens anvisning. Hvis du er i tvivl, skal du kontakte din læge eller sygeplejerske.
Anvendes til voksne oralt
- Profylakse
Hvis opløsningen tages oralt, fortyndes indholdet i ampullen med lidt sukkervand, er den daglige dosis 1½-2 ampuller Ugurol på 500 mg, startes administrationen mindst 1 dag før operationen og fortsætter behandlingen i en periode mindst 3-4 dage efter operationen.
- Terapi
Hvis opløsningen tages oralt, fortyndes indholdet af ampullen i lidt sukkervand, er den daglige dosis 1-2 ampuller Ugurol på 500 mg 3 gange dagligt eller ½-1 ampul Ugurol på 500 mg 6 gange om dagen .
Oral administration er især indiceret:
- i hæmoragiske manifestationer, der opstår i intern medicin, otolaryngologi og tandpleje; til forberedelse af kirurgiske indgreb, hvor det antages, at der kan opstå blødninger fra plasminisk aktivering;
- ved hypermenoré;
- i gynækologiske ædelstene, i blærebetændelse og hæmoragisk proctitis efter strålebehandling for genital carcinom;
- til vedligeholdelse af intravenøs initieret behandling for at forhindre gentagelse af blødning.
Intravenøs brug hos voksne
UGUROL -opløsning gives som en langsom injektion i en vene. Din læge vil beslutte den rigtige dosis til dig, og hvor lang tid du skal tage den.
Brug til voksne til lokal anvendelse
Til lokal anvendelse af UGUROL 500 mg / 5 ml opløsning skal du bruge indholdet af 1 hætteglas og hælde det direkte på blødningsstedet eller anvende det med en tidligere gennemblødt gasbind.
Direkte lokal påføring eller ved hjælp af gaze puder, der tidligere var gennemblødt i opløsningen, er især indiceret til blødning på oro-nasopharyngeal niveau, hvor hurtig hæmostase ønskes.
Brug til børn
Hvis der gives UGUROL -opløsning til et barn fra et års alder, beregnes dosen ud fra barnets vægt. Din læge vil beslutte den rigtige dosis til dit barn, og hvor lang tid du skal tage den.
Brug til ældre
Det er ikke nødvendigt at reducere dosis, medmindre der er påvist nyreinsufficiens.
Anvendes til patienter med nyreproblemer
Hvis du har nyreproblemer, reduceres din dosis tranexaminsyre baseret på en blodprøve (serumkreatininniveau).
Anvendes til patienter med leverproblemer
Det er ikke nødvendigt at reducere dosis.
Indgivelsesmåde
UGUROL -opløsning skal administreres langsomt i en vene
UGUROL -opløsning må ikke injiceres i en muskel.
Overdosering Hvad skal jeg gøre, hvis du har taget for meget Ugurol
Hvis du får mere UGUROL end den anbefalede dosis
Hvis du får mere UGUROL end den anbefalede dosis, kan du have et forbigående fald i blodtrykket. Fortæl det straks til din læge eller apotek.
Hvis du har glemt at tage UGUROL
Tag ikke en dobbeltdosis som erstatning for en glemt dosis.
Spørg din læge eller apotek, hvis du har yderligere spørgsmål om brugen af dette lægemiddel.
Bivirkninger Hvad er bivirkningerne af Ugurol
Ligesom al anden medicin kan denne medicin forårsage bivirkninger, men ikke alle får bivirkninger.
Følgende bivirkninger er blevet observeret med UGUROL:
Almindelig (kan forekomme hos op til 1 ud af 10 patienter)
- virkninger på mave og tarm: kvalme, opkastning, diarré.
Ikke almindelig (kan ramme 1 til 10 brugere ud af 1000)
- virkninger på huden: udslæt
Ikke kendt (hyppigheden kan ikke estimeres ud fra de tilgængelige data)
- utilpashed med hypotension (lavt blodtryk), især hvis injektionen blev givet for hurtigt
- blodpropper;
- virkninger på nervesystemet: kramper
- virkninger på øjnene: synsforstyrrelser, herunder nedsat farvesyn;
- virkninger på immunsystemet: allergiske reaktioner.
Indberetning af bivirkninger
Tal med din læge eller apotek, hvis du får bivirkninger, herunder mulige bivirkninger, der ikke er anført i denne indlægsseddel. Du kan også indberette bivirkninger direkte via det nationale rapporteringssystem på https://www.aifa.gov.it/content/segnalazioni-reazioni-avverse. Ved at rapportere bivirkninger kan du hjælpe med at give mere information om sikkerhed. Af dette lægemiddel.
Udløb og opbevaring
Opbevar denne medicin utilgængeligt for børn
Brug ikke dette lægemiddel efter den udløbsdato, der står på kartonen efter "Udløber den". Udløbsdatoen refererer til den sidste dag i den pågældende måned.
Smid ikke medicin via spildevand eller husholdningsaffald. Spørg din apotek om, hvordan du skal smide medicin, du ikke længere bruger. Dette vil hjælpe med at beskytte miljøet.
UGUROL indeholder
Den aktive ingrediens er tranexamsyre.
Hvert 5 ml hætteglas indeholder 500 mg tranexamsyre. Den anden komponent er vand til injektionsvæsker.
Hvordan UGUROL ser ud og pakningens indhold
UGUROL 500 mg / 5 ml injektionsvæske, opløsning til intravenøs brug, til oral og lokal brug, æske med 5 ampuller
Indlægsseddel: AIFA (Italian Medicines Agency). Indhold offentliggjort i januar 2016. De foreliggende oplysninger er muligvis ikke opdaterede.
For at få adgang til den mest opdaterede version er det tilrådeligt at få adgang til webstedet AIFA (Italian Medicines Agency). Ansvarsfraskrivelse og nyttige oplysninger.
01.0 LÆGEMIDLETS NAVN
UGUROL 500 MG / 5 ML LØSNING TIL INJEKTION TIL INTRAVENØS ANVENDELSE TIL ORAL ELLER LOKAL ANVENDELSE
02.0 KVALITATIV OG KVANTITATIV SAMMENSÆTNING
en 5 ml ampul indeholder: aktiv ingrediens: tranexamsyre 500 mg
Den fulde liste over hjælpestoffer findes i afsnit 6.1
03.0 LÆGEMIDDELFORM
Injektionsvæske, opløsning til intravenøs anvendelse, til oral eller lokal brug.
04.0 KLINISKE OPLYSNINGER
04.1 Terapeutiske indikationer
Forebyggelse og behandling af blødning på grund af generaliseret eller lokal fibrinolyse hos voksne og børn fra et år.
De specifikke indikationer er:
• blødning forårsaget af generaliseret eller lokal fibrinolyse, såsom:
- menorrhagia og metrorrhagia
- gastrointestinal blødning
- urinblødningsforstyrrelser efter prostatakirurgi eller kirurgiske indgreb, der involverer urinvejene
• ENT -kirurgi (adenoidektomi, tonsillektomi, tandekstraktioner);
• gynækologisk kirurgi eller obstetriske lidelser;
• thorax- og mavekirurgi og andre større operationer såsom kardiovaskulær kirurgi;
• behandling af blødninger på grund af administration af et fibrinolytikum.
04.2 Dosering og indgivelsesmåde
Dosering
Voksne
- Oral profylakse
Hvis opløsningen tages oralt, fortyndes indholdet i ampullen med lidt sukkervand, er den daglige dosis 1½-2 ampuller Ugurol på 500 mg, startes administrationen mindst 1 dag før operationen og fortsætter behandlingen i en periode mindst 3-4 dage efter operationen.
- Oral terapi
Hvis opløsningen tages oralt, fortyndes indholdet af ampullen i lidt sukkervand, er den daglige dosis 1-2 ampuller Ugurol på 500 mg 3 gange dagligt eller ½-1 ampul Ugurol på 500 mg 6 gange om dagen .
Oral administration er især indiceret:
- ved hæmoragiske manifestationer, der opstår i intern medicin, otolaryngologi og tandpleje
- til forberedelse af kirurgiske indgreb, hvor det antages, at der kan opstå blødning fra plasminisk aktivering
- ved hypermenoré;
- ved gynækologiske ædelstene, blærebetændelse og hæmoragisk proctitis efter strålebehandling for genital carcinom;
- til vedligeholdelse af behandlinger iværksat intravenøst for at forhindre gentagelse af blødning.
- Intravenøs terapi
Voksne
Medmindre andet er foreskrevet, anbefales følgende doser:
1. standardbehandling af lokal fibrinolyse:
0,5 g (1 x 5 ml ampul) til 1 g (2 x 5 ml ampuller) tranexamsyre ved langsom intravenøs injektion (= 1 ml / minut) to eller tre gange om dagen
2. standardbehandling af generaliseret fibrinolyse:
1 g (2 ampuller á 5 ml) tranexaminsyre ved langsom intravenøs injektion (= 1 ml / minut) hver 6-8 time, svarende til 15 mg / kg legemsvægt.
Nyresvigt
I tilfælde af nyreinsufficiens, som kan indebære en risiko for akkumulering, er brug af tranexamsyre kontraindiceret til patienter med alvorlig nyreinsufficiens (se pkt. 4.3). For patienter med let til moderat nedsat nyrefunktion bør dosis af tranexamsyre reduceres baseret på serumkreatininniveauet.
Leverinsufficiens
Ingen dosisjustering er nødvendig hos patienter med leverinsufficiens.
Ældre borgere
Det er ikke nødvendigt at reducere dosis, medmindre der er påvist nyreinsufficiens.
- Terapi til lokal anvendelse
Indholdet af 1 ampul bruges normalt, som skal hældes direkte på blødningsstedet eller påføres med en tidligere gennemblødt gasbind.
Direkte lokal påføring eller ved hjælp af gaze puder, der tidligere var gennemblødt i opløsningen, er specielt indiceret til blødning på det orale, næsehornsniveau, hvor der ønskes hurtig hæmostase.
Pædiatrisk population
Data om effekt, dosering og sikkerhed for de aktuelt godkendte indikationer som beskrevet i afsnit 4.1 er begrænsede.
- Oral profylakse
Administrer opløsningen oralt i en daglig dosis på 5-10 mg / kg startende administrationen mindst 1 dag før operationen og fortsæt behandlingen i en periode på ikke mindre end 3-4 dage efter operationen.
Hvis opløsningen tages gennem munden, fortyndes hætteglassets indhold med lidt sukkervand.
- Oral terapi
Administrer opløsningen oralt i en dosis på 10-20 mg / kg 3 gange om dagen eller 5-10 mg / kg 6 gange om dagen.
Se "Profylakse" for at tage opløsningen.
- Intravenøs behandling
Hos børn fra et år er dosis for de nuværende godkendte indikationer beskrevet i afsnit 4.1 omkring 20 mg / kg / dag. Der er imidlertid få data om effekt, dosering og sikkerhed for disse indikationer.
Der er ingen udtømmende evalueringer af effekt, dosering og sikkerhed af tranexamsyre hos børn, der gennemgår hjertekirurgi. Aktuelt tilgængelige data er begrænsede og præsenteres i afsnit 5.1.
Indgivelsesmåde
a) Intravenøst
Administration skal nødvendigvis ske ved langsom intravenøs injektion.
b) Oralt
Oralt er opløsningen hovedsageligt indiceret til børn eller patienter med synkebesvær.
c) Til lokal anvendelse
Instruktioner til åbning af hætteglasset
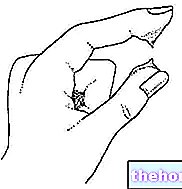
Hætteglassene er udstyret med en sikkerhedsforåbning og skal åbnes som følger:
- placer hætteglasset som angivet i figur 1;
- udøv tryk med tommelfingeren placeret over den FARVEDE prik som vist i figur 2.
04.3 Kontraindikationer
Overfølsomhed over for det aktive stof eller over for et eller flere af hjælpestofferne anført i pkt.6.1.
Akut venøs eller arteriel trombose (se pkt. 4.4).
Fibrinolytiske tilstande på grund af forbrugskoagulopati undtagen i tilfælde, hvor der er dominerende aktivering af det fibrinolytiske system med akut alvorlig blødning (se pkt. 4.4).
Alvorlig nyreinsufficiens (risiko for ophobning).
Anfaldshistorie.
Intratekal og intraventrikulær injektion, intracerebral anvendelse (risiko for cerebralt ødem og kramper).
04.4 Særlige advarsler og passende forholdsregler ved brug
Ovenstående indikationer og indgivelsesmåde skal nøje overholdes:
• intravenøse injektioner skal gives langsomt
• Tranexaminsyre må ikke administreres intramuskulært.
Kramper
Tilfælde af anfald er blevet rapporteret i forbindelse med behandling med tranexamsyre. Ved koronar bypass -transplantation (CABG) kirurgi er de fleste tilfælde forekommet efter intravenøs (IV) injektion af høje doser tranexaminsyre. Med de lavere anbefalede doser af tranexaminsyre, forekomsten af postoperative anfald var den samme som hos ubehandlede patienter.
Synsforstyrrelser
Der bør lægges vægt på mulige synsforstyrrelser, herunder synshandicap, sløret syn, nedsat farvesyn og, om nødvendigt, behandlingen skal stoppes. I tilfælde af langvarig brug af tranexamsyreopløsningen til injektion anbefales regelmæssige oftalmologiske undersøgelser (øjenundersøgelser, herunder synsskarphed, farvesyn, fundus, synsfelt osv.). Ved patologiske oftalmologiske ændringer, især med patologier i nethinden, bør lægen beslutte, efter at have konsulteret en specialist, om behovet for langvarig brug af tranexamsyre, injektionsvæske, opløsning i hvert enkelt tilfælde.
Hæmaturi
I tilfælde af hæmaturi i øvre urinveje er der risiko for urinrørsobstruktion.
Tromboemboliske begivenheder
Inden du bruger tranexaminsyre, skal risikofaktorer for tromboembolisk sygdom overvejes. Tranexamsyreopløsning hos patienter med en historie med tromboembolisk sygdom eller hos dem med en "høj forekomst af tromboemboliske hændelser i" familiehistorien (patienter med høj risiko for trombofili) til injektion bør kun administreres, hvis lægen udtrykkeligt har angivet det efter samråd med en ekspert i hæmostaseologi og under nøje lægeligt tilsyn (se afsnit 4.3).
Tranexaminsyre bør administreres med forsigtighed til patienter, der tager orale præventionsmidler på grund af den øgede risiko for trombose (se pkt. 4.5).
Formidlet intravaskulær koagulation
Patienter med dissemineret intravaskulær koagulation (DIC) kan i de fleste tilfælde ikke behandles med tranexamsyre (se afsnit 4.3) Hvis der træffes beslutning om at administrere tranexamsyre, bør dette kun ske hos patienter, hvor der er en "Overvejende aktivering af fibrinolytisk system med akut alvorlig blødning Normalt nærmer den hæmatologiske profil sig følgende: reduceret lystid for euglobulinproppen; forlænget protrombintid; reducerede plasmaniveauer af fibrinogen, faktorer V og VIII, plasminogen fibrinolysin og alfa-2 macroglobulin; normale plasmaniveauer af protrombinkomplekset, dvs. faktorer II (protrombin), VIII og X; forhøjede plasmaniveauer af fibrinogennedbrydningsprodukter; normalt trombocyttal. Ovenstående forudsætter, at den underliggende sygdom ikke ændrer sig. i sig selv de forskellige elementer i denne profil. I disse akutte normalt en enkelt dosis på 1 g tranexaminsyre eller er det nok til at kontrollere blødningen. Administration af tranexaminsyre i DIC bør kun overvejes, hvis tilstrækkeligt hæmatologisk laboratorieudstyr er tilgængeligt og i nærvær af ekspertpersonale.
04.5 Interaktioner med andre lægemidler og andre former for interaktion
Der er ikke udført interaktionsundersøgelser. Samtidig behandling med antikoagulantia kan kun finde sted under nøje overvågning af en læge med erfaring inden for dette område. Lægemidler, der virker på hæmostase, bør administreres med forsigtighed til patienter behandlet med tranexamsyre. Der er en teoretisk risiko for en potentiel stigning i trombedannelse, som det sker med østrogen. Alternativt kan lægemidlets antifibrinolytiske virkning antagoniseres med trombolytiske lægemidler.
04.6 Graviditet og amning
Kvinder i den fertile alder skal bruge effektive præventionsmidler under behandlingen.
Graviditet
Der er utilstrækkelige kliniske data om anvendelse af tranexamsyre til gravide kvinder. Selvom dyreforsøg ikke rapporterer teratogene virkninger, anbefales det derfor ikke at anvende tranexamsyre i graviditetens første trimester som en sikkerhedsforanstaltning. Begrænsede kliniske data om brug af tranexamsyre ved forskellige blødningstilstande i anden og tredje trimester af graviditeten har ikke rapporteret en skadelig effekt for fosteret.Tranxamsyre kan kun bruges under graviditet, hvis de forventede fordele berettiger den potentielle risiko.
Fodringstid
Tranexaminsyre udskilles i modermælk, derfor anbefales det ikke at amme.
Fertilitet
Der er ingen kliniske data om virkningen af tranexaminsyre på fertiliteten.
04.7 Virkninger på evnen til at føre motorkøretøj og betjene maskiner
Der er ikke udført undersøgelser af evnen til at føre motorkøretøj eller betjene maskiner.
04.8 Bivirkninger
Bivirkninger rapporteret i kliniske forsøg og på grundlag af erfaring efter markedsføring er anført nedenfor efter systemorganklasse.
Tabel med listen over bivirkninger
Bivirkninger rapporteret er inkluderet i nedenstående tabel og er opført efter MedDRA primære systemorganklasse. Inden for hver systemorganklasse er bivirkninger rangeret efter frekvens. Inden for hver frekvenskategori er bivirkninger angivet efter faldende sværhedsgrad. Hyppighedskategorier er defineret som følger: meget almindelig (≥1 / 10); almindelig (≥1 / 100
Indberetning af formodede bivirkninger
Rapportering af formodede bivirkninger, der opstår efter godkendelse af lægemidlet, er vigtig, da det muliggør kontinuerlig overvågning af lægemidlets fordel / risiko -balance. Sundhedspersonale anmodes om at rapportere alle formodede bivirkninger via det nationale rapporteringssystem. "Gadeadresse.
04.9 Overdosering
Der er ikke rapporteret tilfælde af overdosering.
Tegn og symptomer kan være svimmelhed, hovedpine, hypotension og anfald. Anfald har vist sig at forekomme hyppigere ved stigende dosis.
Håndtering af overdosering bør bestå af understøttende pleje.
05.0 FARMAKOLOGISKE EGENSKABER
05.1 Farmakodynamiske egenskaber
Farmakoterapeutisk gruppe: antiblødninger, antifibrinolytika
ATC -kode: B02AA02
Tranexamsyre har en anti-hæmoragisk aktivitet ved at hæmme de fibrinolytiske egenskaber af plasmin.
Der dannes et kompleks, der omfatter tranexaminsyre og plasminogen; tranexamsyre binder til plasminogen, når det omdannes til plasmin.
Tranexaminsyre-plasminkompleksets aktivitet på fibrinaktivitet er lavere end aktiviteten af frit plasmin alene.
Uddannelse in vitro viste, at høje doser tranexaminsyre reducerede komplementaktivitet.
Pædiatrisk population
Børn fra et år:
I litteraturen blev der identificeret 12 effektundersøgelser i pædiatrisk hjertekirurgi, som omfattede 1073 børn, 631 behandlet med tranexamsyre. De fleste af undersøgelserne var placebokontrollerede. Den undersøgte befolkning var heterogen med hensyn til alder, operationstype og doseringsregimer. Resultater fra tranexamsyreundersøgelser indikerer mindre blodtab og mindre behov for blodprodukter ved pædiatrisk hjertekirurgi med hjerte -lunge -bypass (CPB), når der er stor risiko for blødning, især hos cyanotiske patienter eller hos patienter, der gennemgår gentagne kirurgiske indgreb. Den mest passende doseringsplan viste sig at være:
- første bolus på 10 mg / kg efter induktion af anæstesi og før hudincision
- kontinuerlig infusion af 10 mg / kg / t eller injektion i CPB -pumpens grundvæske i en dosis, der passer til CPB -proceduren, eller i henhold til patientens vægt ved en dosis på 10 mg / kg eller i henhold til primingvolumen af pumpens CPB, med den sidste injektion på 10 mg / kg ved afslutningen af den kardiopulmonal bypass -operation.
Selvom undersøgelserne involverede et meget begrænset antal patienter, tyder de få tilgængelige data på, at kontinuerlig infusion er at foretrække, da den bevarer terapeutiske plasmakoncentrationer under hele operationen.
Der er ikke udført specifikke dosis-effekt og farmakokinetiske undersøgelser hos børn.
05.2 Farmakokinetiske egenskaber
Absorption
Højeste plasmakoncentrationer af tranexaminsyre opnås hurtigt efter en kort intravenøs infusion, hvorefter plasmakoncentrationer falder på multi-eksponentiel måde.
Fordeling
Plane-proteinbindingen af tranexaminsyre er cirka 3% ved terapeutiske plasmaniveauer og ser ud til helt at skyldes dens binding til plasminogen Tranexaminsyre binder ikke til serumalbumin. Det indledende distributionsvolumen er cirka 9-12 liter.
Tranexamsyre krydser placenta. Efter administration af en intravenøs injektion af 10 mg / kg til 12 gravide var serumkoncentrationen af tranexamsyre mellem 10 og 53 mcg / ml, mens koncentrationen i navlestrengen i blodet var mellem 4 og 31 mcg / ml. Tranexaminsyre diffunderer hurtigt ind i synovialvæsken og synovialmembranen. Efter administration af en intravenøs injektion på 10 mg / kg til 17 patienter, der blev opereret i knæet, var koncentrationerne i ledvæsken lignende dem, der blev observeret i de relaterede serumprøver. Koncentrationen af tranexaminsyre i en række andre væv svarer til en brøkdel af den, der observeres i blodet (en hundrededel i modermælk, en tiendedel i cerebrospinalvæske, en tiendedel i vandig humor). Tranexamsyre blev påvist i sæd, hvor det hæmmer fibrinolytisk aktivitet, men påvirker ikke sædmigration.
Udskillelse
Det udskilles hovedsageligt i urinen som uændret lægemiddel. Urinudskillelse via glomerulær filtrering er den vigtigste eliminationsvej. Renal clearance er lig med plasmaclearance (110-116 ml / min). Tranexaminsyreudskillelse er cirka 90% i de første 24 timer efter intravenøs administration af 10 mg / kg legemsvægt. Halveringstiden for tranexaminsyre er cirka 3 timer.
Særlige populationer
Plasmakoncentrationer stiger hos patienter med nyreinsufficiens.
Der er ikke udført specifikke farmakokinetiske undersøgelser hos børn.
05.3 Prækliniske sikkerhedsdata
Ikke-kliniske data afslører ingen specifik fare for mennesker baseret på konventionelle undersøgelser af sikkerhedsfarmakologi, toksicitet ved gentagne doser, gentoksicitet, kræftfremkaldende potentiale og reproduktionstoksicitet.
Epileptogen aktivitet blev observeret ved intratekal anvendelse af tranexamsyre hos dyr.
06.0 LÆGEMIDDELOPLYSNINGER
06.1 Hjælpestoffer
Et 500 mg hætteglas med tranexaminsyre / 5 ml indeholder:
vand til injektionsvæsker.
06.2 Uforenelighed
Ingen.
06.3 Gyldighedsperiode
5 år
06.4 Særlige opbevaringsforhold
Ingen
06.5 Den umiddelbare emballages art og emballagens indhold
æske med 5 ampuller à 500 mg / 5 ml
æske med 6 ampuller på 500 mg / 5 ml
06.6 Brugsanvisning og håndtering
07.0 INDEHAVER AF MARKEDSFØRINGSTILLADELSE
Rottapharm S.p.A. - Galleria Unione, 5 - 20122 Milano
08.0 MARKEDSFØRINGSTILLADELSESNUMMER
A.I.C. 021458029 - "500 mg / 5 ml injektionsvæske, opløsning til intravenøs anvendelse, til oral og lokal brug" 5 ampuller
A.I.C. 021458031 - "500 mg / 5 ml injektionsvæske, opløsning til intravenøs anvendelse, til oral og lokal brug" 6 ampuller
09.0 DATO FOR FØRSTE TILLADELSE ELLER FORNYELSE AF TILLADELSEN
Ugurol har været på markedet siden maj 1970 / 31. maj 2005
10.0 DATO FOR REVISION AF TEKSTEN
December 2013