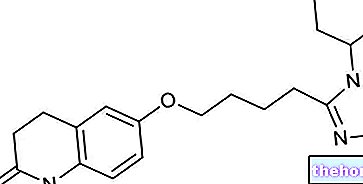
Aktive ingredienser: Paricalcitol
Zemplar 5 mikrogram / ml Injektionsvæske, opløsning
Zemplar indlægssedler er tilgængelige til pakningsstørrelser:- Zemplar 1 mikrogram bløde kapsler
- Zemplar 2 mikrogram Bløde kapsler
- Zemplar 5 mikrogram / ml Injektionsvæske, opløsning
Hvorfor bruges Zemplar? Hvad er det for?
Zemplar er en syntetisk analog af aktivt D -vitamin, der er indiceret til forebyggelse og behandling af høje niveauer af parathyroidhormon i blodet hos patienter med nyreinsufficiens, der gennemgår hæmodialyse. Høje niveauer af parathyroidhormon kan skyldes lave niveauer af "aktivt" D -vitamin hos patienter med nyresvigt.
D -vitamin i sin aktive form sikrer den normale funktion af mange væv i vores krop, herunder nyrer og knogler.
Kontraindikationer Når Zemplar ikke bør bruges
Tag ikke Zemplar
- Hvis du er allergisk (overfølsom) over for paricalcitol eller et af de øvrige indholdsstoffer i Zemplar (se afsnit 6).
- Hvis du har meget høje niveauer af calcium eller D -vitamin i dit blod. Din læge vil overvåge dine blodniveauer og vil kunne informere dig, hvis din sag falder inden for ovenstående betingelser.
Forholdsregler ved brug Det, du skal vide, før du tager Zemplar
Vær særlig forsigtig med Zemplar
- Inden behandlingen påbegyndes, er det vigtigt, at du begrænser mængden af fosfor i din kost. Eksempler på fødevarer med et højt fosforindhold omfatter: te, sodavand, øl, ost, mælk, fløde, fisk, kylling eller oksekødslever, bønner, ærter, korn, nødder og hvede.
- Fosfatbindere, som forhindrer absorption af fosfat fra fødevarer, kan være nødvendige for at kontrollere fosforindholdet.
- Hvis du tager calciumbaserede fosfatbindere, skal din læge muligvis justere din dosis.
- Din læge vil ordinere nogle blodprøver for at overvåge din behandling.
Interaktioner Hvilke lægemidler eller fødevarer kan ændre virkningen af Zemplar
Fortæl det til din læge, sygeplejerske eller apotek, hvis du tager anden medicin eller har brugt det for nylig, også uden recept.
Nogle lægemidler kan påvirke måden Zemplar virker på eller gøre det mere sandsynligt, at du får bivirkninger. Det er især vigtigt at fortælle det til din læge, hvis du tager en af følgende lægemidler:
- medicin til behandling af svampeinfektioner såsom candidiasis eller trost (ketoconazol)
- hjerte- eller blodtryksmedicin (f.eks. digoxin og diuretika eller piller til fjernelse af overskydende vand fra vores krop)
- medicin, der indeholder magnesium (f.eks. nogle fordøjelsesmedicin kaldet antacida, såsom magnesiumtrisilikat)
- lægemidler indeholdende aluminium (f.eks. fosfatbindere, såsom aluminiumhydroxid).
Spørg din læge, sygeplejerske eller apotek til råds, før du tager medicin.
Tager Zemplar sammen med mad og drikke
Zemplar kan tages med eller mellem måltiderne.
Advarsler Det er vigtigt at vide, at:
Graviditet og amning
Hvis du er gravid eller planlægger at blive gravid, skal du fortælle det til din læge eller sygeplejerske, før du tager Zemplar.
Det vides ikke, om Zemplar er sikkert for gravide eller ammende kvinder. Derfor må du ikke tage det, før du har talt med din læge, som vil hjælpe dig med at træffe den bedste beslutning for dig.
Spørg din læge, sygeplejerske eller apotek til råds, før du tager medicin.
Kørsel og brug af maskiner
Der er ikke udført undersøgelser af effekter relateret til evnen til at føre motorkøretøj eller betjene maskiner. Zemplar kan påvirke evnen til at føre køretøjer sikkert eller betjene tunge maskiner. Svimmelhed, træthed og / eller søvnighed er mulige bivirkninger ved behandling med Zemplar.
Kør ikke bil eller brug maskiner, hvis du viser disse symptomer.
Vigtig information om nogle af ingredienserne i Zemplar
Dette lægemiddel indeholder 20% v / v ethanol (alkohol). Hver dosis kan indeholde op til 1,3 g ethanol. Tilstedeværelsen af ethanol i dette lægemiddel er skadeligt for personer med alkoholisme og bør tages behørigt i betragtning, når det administreres til gravide eller ammende kvinder, børn og højrisikogrupper, såsom patienter med leversygdom eller epilepsi.
Dosering og anvendelsesmåde Sådan bruges Zemplar: Dosering
Baseret på resultaterne af laboratorietestene, vil din læge beslutte den passende startdosis for dig. Når behandlingen med Zemplar er startet, er det sandsynligt, at en dosisjustering vil blive foretaget afhængigt af resultaterne af rutinemæssige laboratorietests. Baseret på resultaterne af laboratorietestene vil din læge hjælpe dig med at bestemme den passende dosis Zemplar.
Zemplar vil blive givet til dig af en læge eller sygeplejerske under hæmodialyse gennem blodlinjen, der bruges til at forbinde dig med maskinen. Du behøver ikke en injektion, da Zemplar kan indsættes direkte i det rør, der bruges til din behandling. Zemplar vil blive givet dig hver anden dag, højst tre gange om ugen.
Overdosering Hvad skal jeg gøre, hvis du har taget for meget Zemplar
En overdosis Zemplar kan forårsage unormale stigninger i calcium (i blodet og urinen) og fosfatniveauer i blodet, som kan kræve behandling. Derudover kan en overdosis Zemplar reducere parathyroidhormonniveauer. Symptomer, der kan forekomme kort tid efter at have taget en overdosis Zemplar, omfatter:
- følelse af svaghed og / eller følelsesløshed
- hovedpine
- kvalme eller kvalme
- mundtørhed, forstoppelse
- smerter i muskler eller knogler
- ændring i smag.
Symptomer, der kan forekomme over en længere periode med at tage for meget Zemplar, omfatter:
- mistet appetiten
- døsighed
- vægttab
- ubehag i øjnene
- rhinoré
- kløende hud
- følelse af varme og feber
- tab af libido
- alvorlige mavesmerter
- nyresten
- Blodtrykket kan ændre sig, og der kan forekomme uregelmæssig hjerterytme (hjertebanken).
Zemplar indeholder 30% v / v propylenglycol som hjælpestof. Der har været isolerede rapporter om toksiske virkninger forbundet med administration af høje doser propylenglycol. Sådanne tilfælde bør ikke forekomme, når de administreres til patienter med hæmodialyse, da propylenglycol fjernes fra blodet under dialyseprocessen.
Hvis du har forhøjet calciumindhold i blodet efter at have taget Zemplar, vil din læge sikre, at du får den passende behandling for at genoprette normale calciumniveauer i blodet. Når dit calciumindhold i blodet er vendt tilbage til det normale, vil du sandsynligvis modtage en lavere dosis Zemplar.
Din læge vil stadig kontrollere dit blodniveau, og hvis du bemærker nogen af ovenstående symptomer, skal du straks kontakte en læge.
Bivirkninger Hvad er bivirkningerne af Zemplar
Som al medicin kan Zemplar forårsage bivirkninger, men ikke alle får det.
Forskellige allergiske reaktioner er blevet rapporteret med Zemplar.
Vigtigt: Fortæl det straks til din læge eller sygeplejerske, hvis du bemærker nogen af følgende bivirkninger:
- hvæsen
- åndedræts- eller synkebesvær
- dyspnø
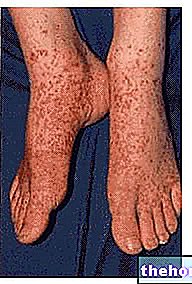
- udslæt, kløende hud eller nældefeber
- hævelse af ansigt, læber, mund, tunge eller hals.
Fortæl det til din læge eller apoteket, hvis du bemærker nogen af følgende bivirkninger:
Mest almindelige bivirkninger (mindst 1 ud af 100 patienter):
- hovedpine
- ændring i smag
- kløende hud
- lave niveauer af parathyroidhormon
- høje calciumniveauer (kvalme eller kvalme, forstoppelse eller forvirring) blodfosfor (sandsynligvis i fravær af symptomer, men med større modtagelighed for brud)
Mindre almindelige bivirkninger (mindst 1 ud af 1.000 patienter):
- allergiske reaktioner (f.eks. hvæsen, hvæsen, udslæt, kløe eller hævelse af ansigt og læber) kløende blærer
- blodinfektioner; reduceret antal røde blodlegemer (anæmi - træthed, åndenød, bleghed) reduceret antal hvide blodlegemer (øget modtagelighed for infektioner); hævede kirtler i nakke, armhule og / eller lyske; forlænget blødningstid (blod størkner ikke let)
- hjerteanfald; slag; brystsmerter; uregelmæssig / hurtig hjerterytme lavt blodtryk (hypotension); forhøjet blodtryk (hypertension);
- koma (dyb bevidstløshed, hvor personen ikke er i stand til at reagere på miljøet)
- usædvanlig træthed, svaghed; svimmelhed besvimelse
- smerter på injektionsstedet
- lungebetændelse (lungeinfektion); væske i lungerne; astma (dyspnø, hoste, åndedrætsbesvær)
- ondt i halsen; kold; feber; influenzalignende symptomer; lyserødt øje (kløende / tørre øjenlåg); øget øjentryk; ørepine; næseblod
- nerve trækninger; forvirring, undertiden alvorlig (delir); uro (angst); nervøsitet personlighedsforstyrrelser (ikke at føle sig som dig selv);
- prikken eller følelsesløshed reduktion af taktile fornemmelser; søvnløshed; nattesved; muskelspasmer i arme og ben, herunder under søvn;
- tør mund; tørst; kvalme; synkebesvær Han trak sig tilbage; mistet appetiten; vægttab; mavepine; diarré og mavesmerter; forstoppelse: anal blødning;
- vanskeligheder med erektion; brystkræft; vaginale infektioner
- brystsmerter; rygsmerter; led / muskelsmerter; følelse af tyngde forårsaget af generaliseret eller lokal hævelse af ankler, fødder og ben (ødem) unormal gangart;
- hårtab; overdreven hårvækst,
- stigning i et leverenzym; forhøjede niveauer af parathyroidhormon; høje niveauer af kalium i blodet reduceret calciumindhold i blodet.
Frekvens ikke kendt:
- hævelse af ansigt, læber, mund, tunge eller hals, hvilket kan forårsage synke- eller vejrtrækningsbesvær kløende hud (nældefeber), maveblødning. Kontakt straks en læge.
Du kan muligvis ikke fortælle, om du har nogen af de ovennævnte bivirkninger, medmindre du fortæller din læge om dem.
Hvis en af bivirkningerne bliver alvorlig, eller hvis du bemærker nogen bivirkninger, der ikke er anført i denne indlægsseddel, skal du straks fortælle det til din læge, sygeplejerske eller apotek.
Udløb og opbevaring
Opbevar denne medicin utilgængeligt for børn.
Dette lægemiddel kræver ingen særlige opbevaringsbetingelser.
Efter åbning skal Zemplar bruges med det samme.
Brug ikke Zemplar efter den udløbsdato, der står på pakningen efter EXP. Udløbsdatoen refererer til den sidste dag i måneden. Brug ikke Zemplar, hvis du bemærker partikler eller uklarhed.
Lægemidler bør ikke bortskaffes via spildevand eller husholdningsaffald. Spørg din apotek om, hvordan du skal smide medicin, du ikke længere bruger. Dette vil hjælpe med at beskytte miljøet.
Andre oplysninger
Hvad Zemplar indeholder
- Den aktive ingrediens er paricalcitol. Hver ml opløsning indeholder 5 mikrogram paricalcitol.
- Øvrige indholdsstoffer er: ethanol (alkohol), propylenglycol og vand til injektionsvæsker.
Hvordan Zemplar ser ud og pakningens indhold
Zemplar injektionsvæske, opløsning er en klar, farveløs, vandig opløsning fri for synlige partikler. Den leveres i pakninger med 5 glasampuller på 1 ml eller 2 ml.
Følgende oplysninger er kun til sundhedspersonale:
Zemplar 5 mikrogram / ml injektionsvæske, opløsning
Klargøring af injektionsvæsken, opløsning Zemplar 5 mikrogram / ml injektionsvæske, opløsning er kun til engangsbrug. Som med alle lægemidler, der administreres ved injektion, skal den fortyndede opløsning undersøges for tilstedeværelse af partikler eller uklarhed inden administration.
Kompatibilitet
Propylenglycol interagerer med heparin og neutraliserer dets virkninger Zemplar injektionsvæske, opløsning indeholder propylenglycol som hjælpestof og skal administreres ad en anden adgangsvej end den, gennem hvilken heparin administreres.
Dette lægemiddel må ikke blandes med andre lægemidler.
Bevaring og gyldighed
Lægemidler, der administreres parenteralt, skal underkastes en "visuel inspektion for tilstedeværelse af korpuskulære stoffer og mulig overskyet tilstand, inden administrationen fortsættes. Opløsningen er klar og farveløs.
Dette lægemiddel kræver ingen særlige opbevaringsbetingelser.
Denne medicin er gyldig i 2 år.
Dosis, metode og tidspunkt for administration
Zemplar injektionsvæske, opløsning skal administreres via en hæmodialyserute.
Voksne
- Startdosis bør beregnes ud fra baseline parathyroidhormon (PTH) niveauer: Startdosis af paricalcitol bør bestemmes ved hjælp af følgende formel:
Startdosis (i mikrogram) = basalt niveau af intakt PTH udtrykt i pmol / l: 8; O = basalt niveau af intakt PTH udtrykt i pg / ml: 80
og bør administreres intravenøst som en bolusdosis, hver anden dag, når som helst under hæmodialysesessionen.
I kliniske forsøg var den maksimale sikre dosis, der blev givet, 40 mikrogram.
- Dosering Titrering:
Det aktuelt accepterede referenceinterval for PTH-niveauer hos dialysepatienter med kronisk nyresvigt i slutstadiet bør ikke overstige 1,5-3 gange den ikke-uremiske øvre grænse på normalt 15,9-31. 8 pmol / l (150-300 pg / ml) for intakt PTH. For at opnå fysiologisk tilstrækkelige resultater skal patienter overvåges nøje og foretages individuel dosisbestemmelse. hypercalcæmi eller et korrigeret, vedvarende forhøjet Ca x P -produkt større end 5,2 mmol2 / l2 (65 mg2 / dl2), dosis bør reduceres eller administrationen afbrydes, indtil disse parametre er vendt tilbage til det normale., skal paricalcitol genindgives ved en lavere dosis.Dosis af paricalcitol skal muligvis reduceres, når PTH-niveauer falder som reaktion på terapi.
Følgende tabel giver et eksempel på en anbefalet fremgangsmåde til bestemmelse af doseringen:
Indlægsseddel: AIFA (Italian Medicines Agency). Indhold offentliggjort i januar 2016. De foreliggende oplysninger er muligvis ikke opdaterede.
For at få adgang til den mest opdaterede version er det tilrådeligt at få adgang til webstedet AIFA (Italian Medicines Agency). Ansvarsfraskrivelse og nyttige oplysninger.
01.0 LÆGEMIDLETS NAVN
ZEMPLAR INJEKTERBAR LØSNING
02.0 KVALITATIV OG KVANTITATIV SAMMENSÆTNING
Zemplar 2 mcg / ml injektionsvæske, opløsning:
Hver ml injektionsvæske, opløsning indeholder 2 mcg paricalcitol.
Hvert 1 ml hætteglas indeholder 2 mcg paricalcitol.
Hvert 1 ml hætteglas indeholder 2 mcg paricalcitol.
Zemplar 5 mcg / ml injektionsvæske, opløsning:
Hver ml injektionsvæske, opløsning indeholder 5 mcg paricalcitol.
Hvert 1 ml hætteglas indeholder 5 mcg paricalcitol.
Hvert 2 ml hætteglas indeholder 10 mcg paricalcitol.
Hvert 1 ml hætteglas indeholder 5 mcg paricalcitol.
Hvert 2 ml hætteglas indeholder 10 mcg paricalcitol.
Hjælpestoffer: Ethanol (20% v / v) og propylenglycol (30% v / v)
Den fulde liste over hjælpestoffer findes i afsnit 6.1.
03.0 LÆGEMIDDELFORM
Injicerbar løsning
Klar og farveløs vandig opløsning uden synlige partikler.
04.0 KLINISKE OPLYSNINGER
04.1 Terapeutiske indikationer
Paricalcitol er indiceret til voksne til forebyggelse og behandling af sekundær hyperparatyreoidisme hos patienter med kronisk nyresvigt, der gennemgår hæmodialyse.
04.2 Dosering og indgivelsesmåde
Dosering
Voksne
1) Startdosis bør beregnes ud fra basalniveauerne af parathyreoideahormon (PTH):
Startdosis af paricalcitol bør bestemmes ved hjælp af følgende formel:
ELLER
og bør administreres intravenøst som en bolusdosis, med en maksimal frekvens hver anden dag, når som helst under hæmodialysesessionen.
I kliniske forsøg var den maksimale sikre dosis, der blev givet, 40 mikrogram.
2) Dosering titrering:
Det aktuelt accepterede referenceinterval for PTH-niveauer hos dialysepatienter med kronisk nyresvigt i slutstadiet bør ikke overstige 1,5-3 gange den ikke-uremiske øvre grænse på normalt 15,9-31. 8 pmol / l (150-300 pg / ml) for intakt PTH.
For at opnå fysiologisk tilstrækkelige resultater skal patienter overvåges omhyggeligt og individuel dosistitrering udføres.
Hvis der konstateres hypercalcæmi eller et korrigeret, vedvarende forhøjet Ca x P -produkt større end 5,2 mmol2 / l2 (65 mg2 / dl2), skal dosis reduceres eller administrationen afbrydes, indtil disse parametre ikke vil blive inkluderet i standarden. Derefter skal paricalcitol genindgives ved en lavere dosis. Paricalcitol -doseringen skal muligvis reduceres, når PTH -niveauer falder som reaktion på terapi.
Følgende tabel giver et eksempel på en anbefalet fremgangsmåde til dosistitrering:
Når paricalcitoldoseringen er fastslået, skal serumkalcium- og fosfatniveauer måles mindst en gang om måneden. Overvågning af intakt serum PTH hver tredje måned anbefales.
Under paricalcitols dosisjusteringsfase kan det være nødvendigt at udføre laboratorietest oftere.
Nedsat leverfunktion
Gratis paricalcitolkoncentrationer hos patienter med let til moderat nedsat leverfunktion svarer til dem, der findes hos raske forsøgspersoner, og dosisjustering er ikke nødvendig i denne patientpopulation. Der er stadig ingen erfaring med patienter med svært nedsat leverfunktion.
Pædiatrisk population (0-18 år)
Sikkerhed og virkning af Zemplar er ikke fastslået hos børn. Der er ingen data tilgængelige for børn under 5 år. Aktuelt tilgængelige data om pædiatriske patienter er beskrevet i afsnit 5.1, men der kan ikke gives nogen anbefaling om dosering.
Ældre patienter (> 65 år)
Erfaring med patienter 65 år og ældre, der fik paricalcitol i fase III -undersøgelser, er noget begrænset. Under disse undersøgelser blev der ikke observeret væsentlige forskelle i lægemidlets effekt eller sikkerhed mellem patienter 65 år og ældre og yngre patienter.
Indgivelsesmåde
Zemplar injektionsvæske, opløsning skal administreres via en hæmodialyserute.
04.3 Kontraindikationer
Overfølsomhed over for det aktive stof eller over for et eller flere af hjælpestofferne anført i pkt.6.1.
D -vitamin toksicitet
Hypercalcæmi.
04.4 Særlige advarsler og passende forholdsregler ved brug
En overdreven hæmning af parathyroidhormonsekretion kan forårsage en stigning i serumkalciumniveauer og kan føre til begyndelsen af osteo-metabolisk sygdom. For at opnå tilstrækkelige fysiologiske referenceværdier skal patienter overvåges omhyggeligt og individuel dosistitrering udføres.
Hvis der opstår klinisk signifikant hypercalcæmi, og patienten behandles med en calciumbaseret phosphationchelator, bør dosis af denne chelator reduceres, eller administrationen afbrydes.
Kronisk hypercalcæmi kan være forbundet med generaliserede vaskulære forkalkninger og andre forkalkninger af blødt væv.
Phosphat eller vitamin D-relaterede lægemidler bør ikke tages samtidigt med paricalcitol, da der kan være en øget risiko for hypercalcæmi, og der kan forekomme en stigning i Ca x P-produkt (se pkt. 4.5).
Digitalis toksicitet forstærkes af hypercalcæmi af enhver oprindelse; derfor bør der udvises ekstrem forsigtighed hos patienter, der får paricalcitolbehandling, der tager samtidig digitalis (se pkt. 4.5).
Der skal udvises forsigtighed, hvis paricalcitol og ketoconazol administreres samtidigt (se pkt. 4.5).
Denne medicin indeholder 20% v / v ethanol (alkohol). Hver dosis kan indeholde op til 1,3 g ethanol. Tilstedeværelsen af ethanol i dette lægemiddel kan være skadelig for personer, der lider af alkoholisme og skal tages i betragtning, når det administreres til gravide og ammende kvinder, pædiatriske personer og højrisikogrupper, f.eks. Patienter med leversygdom eller epilepsi.
04.5 Interaktioner med andre lægemidler og andre former for interaktion
Der er ikke udført interaktionsundersøgelser med paricalcitol i injicerbar form. Imidlertid blev der udført en undersøgelse for at evaluere interaktionen mellem ketoconazol og paricalcitol ved hjælp af kapselformuleringen.
Fosfatprodukter eller D -vitaminanaloger bør ikke tages samtidig med paricalcitol på grund af en øget risiko for hypercalcæmi og en stigning i CaxP -produktet (se pkt. 4.4).
Administration af høje doser af calciumholdige lægemidler eller thiaziddiuretika kan øge risikoen for hypercalcæmi.
Lægemidler indeholdende aluminium (f.eks. Antacida eller phosphationchelatorer) bør ikke indgives i langtidsbehandling i kombination med lægemidler indeholdende D-vitamin, da dette kan resultere i øgede blodniveauer af aluminium og knogletoksicitet fra aluminium.
Lægemidler indeholdende magnesium (f.eks. Antacida) bør ikke tages samtidigt med medicin, der indeholder D -vitamin, da der kan forekomme hypermagnesæmi.
Ketoconazol er kendt for at være en ikke-specifik hæmmer af flere cytochrom P450-enzymer.
Tilgængelige in vivo- og in vitro -data tyder på, at ketoconazol kan interagere med enzymer, der er ansvarlige for metabolismen af paricalcitol og andre D -vitaminanaloger.
Der bør udvises særlig forsigtighed, når paricalcitol administreres samtidigt med ketoconazol (se pkt. 4.4). Virkningen af flere doser ketoconazol administreret i en dosis på 200 mg, to gange dagligt (BID) i 5 dage, på paricalcitolkapslers farmakokinetik blev undersøgt hos raske forsøgspersoner.I nærvær af ketoconazol blev Cmax for paricalcitol påvirket i middelværdien halveringstiden for paricalcitol var 17,0 timer i nærvær af ketoconazol sammenlignet med 9,8 timer, da paricalcitol blev administreret alene. Resultaterne af denne undersøgelse indikerer, at efter oral administration af paricalcitol den maksimale stigning i AUCo-? Af paricalcitol på grund af lægemiddelinteraktion med ketoconazol ikke bør være mere end to gange.
Digitalis toksicitet forstærkes af tilstedeværelsen af hypercalcæmi af enhver oprindelse; derfor bør der udvises ekstrem forsigtighed, hvis digitalis ordineres samtidigt med paricalcitol (se pkt. 4.4).
04.6 Graviditet og amning
Graviditet
Der er utilstrækkelige data om brugen af paricalcitol til gravide Dyrestudier har vist reproduktionstoksicitet (se pkt. 5.3) Den potentielle risiko for mennesker er ukendt. Zemplar bør ikke bruges under graviditet, medmindre det er klart nødvendigt.
Amning
Dyreforsøg har vist, at paricalcitol eller dets metabolitter udskilles i modermælk i små mængder. Der skal træffes en beslutning om, hvorvidt amning skal afbrydes, eller om parikalcitolbehandling skal afbrydes under hensyntagen til fordelen ved amning for barnet og fordelen ved paricalcitolbehandling for kvinden.
04.7 Virkninger på evnen til at føre motorkøretøj og betjene maskiner
Paricalcitol påvirker ubetydeligt evnen til at føre motorkøretøj eller betjene maskiner. Svimmelhed kan forekomme efter administration af paricalcitol (se pkt.4.8).
04.8 Bivirkninger
I fase II / III / IV kliniske forsøg blev cirka 600 patienter behandlet med Zemplar. Samlet set rapporterede 6% af patienterne behandlet med Zemplar bivirkninger.
Den mest almindelige bivirkning forbundet med Zemplar-behandling var hypercalcæmi, der forekom hos 4,7% af patienterne. Hypercalcæmi er afhængig af niveauet af parathyroidhormonover undertrykkelse og kan minimeres ved tilstrækkelig dosistitrering.
Mulige bivirkninger relateret til paricalcitol, både klinisk og laboratorium, er anført i følgende tabel i henhold til MedDRA -konventionen efter systemorganklassificering og frekvens. Med hensyn til frekvens blev følgende kategorier brugt: Meget almindelig (≥ 1 / 1O); almindelig (≥ 1/100,
Indberetning af formodede bivirkninger
Rapportering af formodede bivirkninger, der opstår efter godkendelse af lægemidlet, er vigtig, da det giver mulighed for løbende overvågning af lægemidlets fordel / risiko -sundhedspersonale bedes rapportere eventuelle bivirkninger via det nationale rapporteringssystem. Adresse www.agenziafarmaco.gov .it/it/responsabili.
04.9 Overdosering
Der er ikke rapporteret tilfælde af overdosering.
Overdosering af Paricalcitol kan føre til hypercalcæmi, hypercalcinuri, hyperphosphatæmi og overdreven PHT -undertrykkelse (se pkt. 4.4).
I tilfælde af overdosering skal tegn og symptomer på hypercalcæmi (serumcalciumniveauer) overvåges og rapporteres til lægen. Behandlingen skal startes korrekt.
Paricalcitol elimineres ikke signifikant ved dialyse.Behandling af patienter med "klinisk signifikant hypercalcæmi er" øjeblikkelig dosisreduktion eller øjeblikkelig afbrydelse af paricalcitolbehandling og en diæt med lavt indhold af calcium, suspension af calciumtilskud, patientmobilisering, kontrol af elektrolyt og væskeubalancer, en vurdering af ændringer i det elektrokardiografiske spor (af grundlæggende betydning hos patienter, der behandles med digitalis), og "hæmodialyse eller dialyse peritoneal med calciumfrit dialysat, som tilladt.
Når serumcalciumniveauet er vendt tilbage til normale grænser, kan paricalcitol genindgives ved en lavere dosis. Hvis der opstår en vedvarende og markant stigning i serumkalciumniveauer, bør der tages højde for de forskellige tilgængelige terapeutiske alternativer. Disse omfatter brug af lægemidler som fosfater og kortikosteroider samt foranstaltninger til at fremkalde diurese.
Zemplar injektionsvæske, opløsning indeholder 30% volumenprocent propylenglycol som hjælpestof. Isolerede tilfælde af centralnervesystemdepression, hæmolyse og laktatacidose er blevet rapporteret som en toksisk virkning forbundet med administration af høje doser propylenglycol. Selvom sådanne toksiske virkninger ikke forventes at forekomme efter administration af Zemplar, da propylenglycol elimineres under dialyseprocessen, skal risikoen for toksiske virkninger i tilfælde af overdosering stadig overvejes.
05.0 FARMAKOLOGISKE EGENSKABER
05.1 Farmakodynamiske egenskaber
Farmakoterapeutisk klassifikation: Antiparathyroidea -midler, ATC -kode: H05BX02
Handlingsmekanisme
Paricalcitol er en syntetisk analog af calcitriol, den biologisk aktive form for D-vitamin, med ændringer på sidekæden (D2) og på A (19-nor) ringen. I modsætning til calcitriol er paricalcitol en selektiv aktivator af vitamin D (VDR) Paricalcitol stimulerer selektivt D -vitaminreceptorer i biskjoldbruskkirtlerne uden at forårsage en stigning i D -vitaminreceptorer i tarmen og er mindre aktiv på knogleresorption. Desuden stimulerer paricalcitol de calciumfølsomme receptorer (CaSR), der er til stede i biskjoldbruskkirtlerne. Følgelig reducerer paricalcitol niveauerne af parathyreoideahormon (PTH) ved at hæmme parathyroidea -proliferation og reducere PTH -syntese og sekretion, med minimal indvirkning på calcium- og fosforindholdet; paricalcitol kan virke direkte på osteoblaster for at bevare knoglevolumen og forbedre mineraliseringsoverflader. Korrektion af ændrede niveauer af parathyroidhormon kan sammen med normalisering af calcium og fosforhomeostase forhindre eller helbrede metabolisk knoglesygdom forbundet med kronisk nyresvigt.
Pædiatrisk population
Sikkerhed og effekt af Zemplar blev evalueret i et 12-ugers, randomiseret, dobbeltblindet, placebokontrolleret studie med 29 hæmodialyse pædiatriske patienter med kronisk nyresvigt i slutstadiet i alderen 5 til 19 år. I undersøgelsen behandlede de seks yngste patienter med Zemplar varierede i alderen fra 5 til 12 år. Startdosis af Zemplar var henholdsvis 0,04 mcg / kg 3 gange om ugen, hvis iPTH -niveauer ved baseline var under 500 pg / ml eller 0,08 mcg / kg 3 gange om ugen, hvis baseline -niveauer var på iPTH var ≥ 500 pg / ml. Dosen af Zemplar blev justeret i trin på 0,04 mcg / kg baseret på serumniveauer af iPTH, calcium og Ca x P -produkt. 67% af patienterne behandlet med Zemplar og 14% af de behandlede patienter afsluttede undersøgelsen med placebo. 60% af forsøgspersonerne i Zemplar -gruppen havde 2 på hinanden følgende 30% fald i iPTH -niveauer fra baseline sammenlignet med 21% af patienterne i placebogruppen. På grund af overdrevne stigninger i iPTH -niveauer måtte 71% af patienterne i placebogruppen droppe studiet. Hverken i Zemplar -gruppen eller i placebogruppen udviklede hypercalcæmi.Der er ingen data tilgængelige for patienter under 5 år.
05.2 Farmakokinetiske egenskaber
Fordeling
Paricalcitols farmakokinetik er undersøgt hos patienter med kronisk nyresvigt (CRF), som krævede hæmodialyse. Paricalcitol administreres som en intravenøs bolusinjektion. Inden for to timer efter administration af doser fra 0,04 og 0,24 mcg / kg faldt paricalcitolkoncentrationer hurtigt; efterfølgende faldt paricalcitolkoncentrationer på en logaritmisk lineær måde med en gennemsnitlig halveringstid på cirka 15 timer. Derudover blev der ikke observeret nogen ophobning af paricalcitol i nærvær af flere doser. Plasmaproteinbindingen af paricalcitol in vitro det viste sig at være omfattende (> 99,9%) og ikke-mætteligt i hele koncentrationsområdet mellem 1 ng / ml og 100 ng / ml.
Biotransformation
Både i urin og fæces er flere ukendte metabolitter blevet identificeret, og der er ikke fundet påviselig paricalcitol i urinen. Disse metabolitter er ikke blevet karakteriseret eller identificeret. Samlet set bidrog disse metabolitter til 51% af urinradioaktivitet og 59% til fækal radioaktivitet.
Eliminering
Hos raske forsøgspersoner blev der udført en undersøgelse, hvor en enkelt bolusdosis på 0,16 mcg / kg 3H-paricalcitol (n = 4) blev administreret intravenøst, radioaktiviteten observeret i plasma var Paricalcitol elimineret hovedsagelig ved hepatobiliær udskillelse, da 74% af den radioaktive dosis blev genfundet i fæces, og kun 16% blev genfundet i urinen.
Særlige populationer
Køn, race og alder: Hos voksne undersøgte patienter blev der ikke observeret nogen alders- eller kønsrelaterede farmakokinetiske forskelle Ingen farmakokinetiske forskelle på grund af race blev identificeret.
Nedsat leverfunktion: Gratis paricalcitolkoncentrationer hos patienter med let til moderat nedsat leverfunktion svarer til dem, der er rapporteret hos raske personer, og der er ikke behov for dosisjustering i denne patientpopulation. Der er ingen erfaring med patienter med svært nedsat leverfunktion.
05.3 Prækliniske sikkerhedsdata
De fremtrædende data fra toksicitetsundersøgelser ved gentagne doser hos gnavere og hunde blev generelt tilskrevet den calcemiske aktivitet af paricalcitol. Virkninger, der ikke var klart relateret til hypercalcæmi, omfattede et fald i antallet af hvide blodlegemer hos hunde., Forekomsten af thymisk atrofi hos hunde og tilstedeværelsen af ændrede værdier af aktiveret partiel thromboplastintid (øget hos hunde og faldet hos rotter) Der blev ikke observeret ændringer i antallet af hvide blodlegemer i kliniske undersøgelser.
Paricalcitol forårsagede ikke negative virkninger på rotters fertilitet, og det blev vist, at det ikke besidder nogen teratogen aktivitet hos hverken rotter eller kaniner. Høje doser af andre D -vitaminpræparater indgivet til drægtige dyr inducerede teratogenese.
Paricalcitol har vist sig at påvirke fostrets levedygtighed og kan fremme en signifikant stigning i perinatal og postnatal dødelighed hos nyfødte rotter, når det administreres i toksiske doser hos moderen.
Under en række genetiske toksicitetstests in vitro og in vivo, har det vist sig, at paricalcitol ikke besidder nogen potentiel genotoksisk aktivitet.
Undersøgelser af kræftfremkaldende egenskaber hos gnavere angiver ikke tilstedeværelsen af nogen særlig risiko, når paricalcitol anvendes til mennesker.
Doser administreret og / eller systemisk eksponering for paricalcitol er lidt højere end terapeutiske doser / systemiske eksponeringer.
06.0 LÆGEMIDDELOPLYSNINGER
06.1 Hjælpestoffer
Ethanol (20% v / v)
Propylenglycol
Vand til injektionsvæsker
06.2 Uforenelighed
I mangel af kompatibilitetsundersøgelser må dette lægemiddel ikke blandes med andre lægemidler.
Propylenglycol interagerer med heparin og neutraliserer dets virkninger Zemplar injektionsvæske, opløsning indeholder propylenglycol som hjælpestof og skal administreres ad en anden adgangsvej end den, gennem hvilken heparin administreres.
06.3 Gyldighedsperiode
2 år.
Anvendes umiddelbart efter åbning.
06.4 Særlige opbevaringsforhold
Denne medicin kræver ingen særlige opbevaringsbetingelser.
06.5 Den umiddelbare emballages art og emballagens indhold
Zemplar 2 mcg / ml injektionsvæske, opløsning:
Hvert hætteglas af type 1 indeholder 1 ml injektionsvæske, opløsning.
Hvert hætteglas af type 1 indeholder 1 ml injektionsvæske, opløsning.
Zemplars præsentationer er:
En pakning indeholdende 5 ampuller på 1 ml injektionsvæske, opløsning.
En pakning indeholdende 5 hætteglas med 1 ml injektionsvæske, opløsning.
Zemplar 5 mcg / ml injektionsvæske, opløsning:
Hvert hætteglas af type 1 indeholder 1 ml eller 2 ml injektionsvæske, opløsning.
Hvert hætteglas af type 1 indeholder 1 ml eller 2 ml injektionsvæske, opløsning.
Zemplars præsentationer er:
En pakning indeholdende 5 ampuller på 1 ml injektionsvæske, opløsning.
En pakning indeholdende 5 ampuller med 2 ml injektionsvæske, opløsning.
En pakning indeholdende 5 hætteglas med 1 ml injektionsvæske, opløsning.
En pakning indeholdende 5 hætteglas med 2 ml injektionsvæske, opløsning.
Ikke alle pakningsstørrelser er nødvendigvis markedsført.
06.6 Brugsanvisning og håndtering
Lægemidler, der administreres parenteralt, skal underkastes en "visuel inspektion for tilstedeværelse af korpuskulære stoffer og mulig overskyet tilstand, inden administrationen fortsættes. Opløsningen er klar og farveløs.
Kun til engangsbrug. Ubrugt medicin og affald fra denne medicin skal bortskaffes i overensstemmelse med lokale regler.
07.0 INDEHAVER AF MARKEDSFØRINGSTILLADELSE
AbbVie S.r.l.
S.R. 148 Pontina km 52 snc
04011 Campoverde di Aprilia (LT)
08.0 MARKEDSFØRINGSTILLADELSESNUMMER
Zemplar 2 mcg / ml injektionsvæske, opløsning "5 ampuller à 1 ml - AIC n. 036374128
Zemplar 2 mcg / ml injektionsvæske, opløsning "5 hætteglas med 1 ml - AIC n. 036374155
Zemplar 5 mcg / ml injektionsvæske, opløsning "5 ampuller på 1 ml - AIC n. 036374015
Zemplar 5 mcg / ml injektionsvæske, opløsning "5 ampuller à 2 ml - AIC n. 036374027
Zemplar 5 mcg / ml injektionsvæske, opløsning "5 hætteglas med 1 ml - AIC n. 036374130
Zemplar 5 mcg / ml injektionsvæske, opløsning "5 hætteglas med 2 ml - AIC n. 036374142
09.0 DATO FOR FØRSTE TILLADELSE ELLER FORNYELSE AF TILLADELSEN
Dato for første godkendelse: 22. januar 2005
Dato for seneste fornyelse: 22. november 2010
10.0 DATO FOR REVISION AF TEKSTEN
09/2016