Denne sygdom skylder sit navn til den amerikanske endokrinolog, der opdagede den: Frederic Crosby Bartter.Den årlige forekomst er anslået til 1 / 830.000.
Der er flere varianter af Bartters syndrom, hvis transmission, selvom den stadig er autosomal, kan variere fra recessiv til dominerende afhængigt af sagen.
Hvis det ikke straks diagnosticeres og behandles, kan Bartters syndrom alvorligt kompromittere patientens udvikling, vækst og livskvalitet. Desuden reduceres levetiden i særligt alvorlige tilfælde betydeligt.
Bemærk venligst
Bartters syndrom bør IKKE forveksles med Schwartz-Bartters syndrom, en sygdom præget af "nedsat sekretion af antiduiretisk hormon (ADH), også kendt som syndromet med upassende ADH-sekretion (SIADH).
.som forekommer på niveauet for Henles sløjfe, kan tilskrives en "ændring af syntesen af nogle kanal- / transportørreceptorer (særlige proteiner, der transporterer ioner af forskellig art) placeret i dette" område af nyrerne. Dette fænomen er forårsaget ved en række genetiske mutationer, der påvirker generne, der koder for de førnævnte særlige proteiner.
De forskellige varianter af Bartters syndrom skelnes efter det berørte gen. Flere detaljerede oplysninger om dette findes i det følgende kapitel.
.
Følgende tabel viser derfor de forskellige varianter af syndromet, de involverede muterede gener, proteinerne (kanalreceptorer / transportører), som de koder for, og den kliniske præsentation af den pågældende variant.
Variant
Muteret gen
Kanal / transportør involveret
Klinisk præsentation
Bartters syndrom type I
Gene SLC12A1
NKCC2 (natrium-kalium-chlortransportør eller Na + / K + / 2Cl-)
Prænatal (eller infantil) Bartters syndrom
Bartters syndrom type II
Gene KCNJ1
ROMK (kaliumkanal i nyrens ydre medulla)
Prænatal (eller infantil) Bartters syndrom
Bartters syndrom type III
Gene CLNKb
CLCNKb (Kb -type klorkanal)
Klassisk Bartters syndrom
Bartters syndrom type IV eller IV A
Gene BSND
Barttina (beta -underenhed af klorkanaler af Ka- og Kb -type)
Prænatal (eller infantil) Bartters syndrom og sensorineural høretab
Bartters syndrom type IV B
CLCNKa og CLCNKb gener
CLCNKa (klorkanal af Ka -typen) og CLCNKb
Prænatal (eller infantil) Bartters syndrom og sensorineural døvhed
Type V Bartters syndrom
CASR -gen
CaSR (calciumfølsom receptor)
Bartters syndrom med hypokalcæmi
Som det fremgår af tabellen, er det på trods af tilstedeværelsen af fem genetiske varianter ikke muligt at skelne så mange kliniske former; faktisk skelnes kun fire: prænatal eller infantil Bartters syndrom (type I og II), klassisk Bartters syndrom (type III), prænatal eller infantil Bartters syndrom forbundet med sensorineural døvhed (type IV A og IV B; nogle kilder dog de grupperer disse varianter sammen med type I og II) og endelig Bartters syndrom med hypokalcæmi (type V).
Vidste du, at ...
I betragtning af eksistensen af en variant IV (eller IV A) og en variant IV B af Bartters syndrom overvejer nogle kilder samlet set seks varianter af Bartters syndrom. Andre kilder betragter dog variant IV B som en undertype af variant IV og af denne grund overveje eksistensen af kun fem genetiske varianter af Bartters syndrom.
Varianter af type I, II, III, IV og IV B er autosomale recessivt overførte sygdomme, hvilket betyder, at individet for at manifestere syndromet skal have begge muterede alleler, der arver dem fra de forældre, der derfor vil være sunde bærere. af syndromet er derimod en autosomal dominerende transmissionssygdom, hvilket betyder, at for at manifestere symptomerne er det tilstrækkeligt, at patienten har en enkelt muteret allel, som derfor kan arves selv af kun én (også syg ) af de to forældre.
Bartters pseudo-syndrom
Bartters pseudo-syndrom er en tilstand, der er karakteriseret ved symptomer, der ligner dem, der fremkaldes af Bartters syndrom, men hvis årsag findes ved misbrug af vanddrivende medicin såsom furosemid.
Gitelman syndrom
Dette syndrom er forårsaget af en lokal mutation på SLC12A3-genet, der koder for natriumchlor-cotransporter (NCC). På grund af denne mutation - transmitteret på en autosomal recessiv måde - undergår patienten en forringelse af reabsorptionen af natrium, chlor og kalium på niveau med den distale krølle tubuli, i modsætning til Bartters syndrom, hvor nedsættelsen af resorption er lokaliseret i "Dog , Gitelmans syndrom kan give anledning til symptomer svarende til Bartters syndrom, hvorfor det i klinisk praksis nogle gange kan være svært at skelne mellem de to sygdomme.
, hypochloræmi og metabolisk alkalose, som kan være forbundet med hyperreninæmi (højt blodrenin) og hyperaldosteronisme. Det er klart, at alle disse tilstande igen kan give anledning til en række symptomer, der kan kompromittere patientens livskvalitet (f.eks. Kvalme, opkastning, svimmelhed, svaghed, hovedpine, hypotension osv.).
Ud over det, der er blevet sagt hidtil, kan hver variant give anledning til specifikke manifestationer og symptomer, der er tæt forbundet med det muterede gen og til den deraf følgende involvering af kanalen eller cotransportøren, som dette gen koder for. Derfor vil de typiske symptomer og manifestationer forbundet med hver af de fem forskellige former for Bartters syndrom kort blive beskrevet nedenfor.
Bartters syndrom type I
Ved Bartters syndrom type I påvirker mutationerne genet, der koder for natrium-kalium-klor-transportøren, der er til stede på Henles sløjfe. På grund af den kompromitterede reabsorption opstår hypovolæmi på grund af tab af salte. På samme tid, da genoptagelse af calcium også er knyttet til aktiviteten af den førnævnte cotransporter, er vi vidne til begyndelsen af hypercalciuri. Alt dette kan føre til begyndelsen af nefrocalcinose. Det er også muligt at opleve hypermagnesuri. Polyhydramnios sekundært til føtal polyuri kan udvikle sig i prænatalperioden.
Bartters syndrom type II
Bartters syndrom type II er forårsaget af en mutation i genet, der koder for adrenal medulla's kaliumkanal. Manifestationer og symptomer ligner dem i variant I, og også i dette tilfælde kan man støde på polyhydramnios sekundært til føtal polyuri. På et tidligt tidspunkt kan den nyfødte dog opleve forbigående hyperkalæmisk metabolisk acidose. Denne tilstand udvikler sig derefter mod det karakteristiske kliniske billede af Bartters syndrom.
Bartters syndrom type III
Også kendt som klassisk Bartters syndrom, er variant III af sygdommen forårsaget af mutationer i genet, der koder for klorkanalen af Kb -typen. Da klorkanaler af Ka -typen bevares i denne form, har symptomerne en tendens til at være lettere, selvom de stadig er til stede. Der er generelt ingen nefrocalcinose.
Bartters syndrom type IV og IV B
I begge typer af variant IV er der involvering af gener involveret i den korrekte syntese af Ka- og Kb -klorkanalerne. Da begge kanaler er kompromitteret, har symptomerne en tendens til at være mere alvorlige end for variant III af syndromet. Spædbørn kan i første omgang vise et klinisk billede, der efterligner hypoaldosteronisme, men som derefter udvikler sig mod hypokalæmisk metabolisk alkalose, når kroppen forsøger at kompensere for manglen på aktivitet i de førnævnte calciumkanaler. Karakteristisk for varianter IV og IV B af Bartters syndrom er udseendet af sensorineural døvhed.
Type V Bartters syndrom
Variant V af Bartters syndrom er forårsaget af en mutation, der påvirker genet, der koder for den calciumfølsomme receptor, involveret i hæmning af reabsorptionen af vand og forskellige ioner, såsom calcium, kalium og natrium. Receptor fører til udseende af hypocalcæmi og deraf følgende hypercalciuri forbundet med de karakteristiske symptomer på Bartters syndrom.
Vidste du, at ...
Varianter I, II, IV og IV B af Bartters syndrom - samt med navnet på prænatal Bartters syndrom - kaldes undertiden også som hypeprostaglandin E2 syndrom, da de er karakteriseret ved en stigning i plasmaniveauer af dette prostaglandin.
- har til formål at identificere tilstedeværelse og koncentration af elektrolytter (natrium, kalium, chlorid, magnesium, bikarbonat, calcium) og specifikke stoffer (renin og aldosteron) i plasma og / eller urin.Den endelige diagnose er imidlertid kun mulig ved udførelse af specifikke genetiske tests.
Differentialdiagnosen skal derimod placeres mod Bartters pseudosyndrom, Gitelmans syndrom, cystisk fibrose og cøliaki.
I tilfælde, hvor der er en vis risiko (for eksempel forældre med raske og / eller syge bærere) for, at den nyfødte kan manifestere sygdommen, er prænatal diagnose også mulig.
fra:
- Tilskud af mineralsalte (især men ikke udelukkende kalium) for at kompensere for den manglende reabsorption;
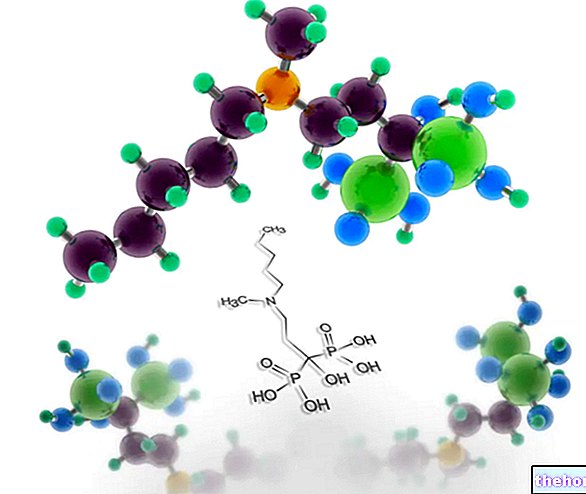
- Ikke-steroide antiinflammatoriske lægemidler (NSAID'er) såsom f.eks. Indomethacin Disse lægemidler administreres med det formål at reducere alt for høje niveauer af prostaglandin E2;
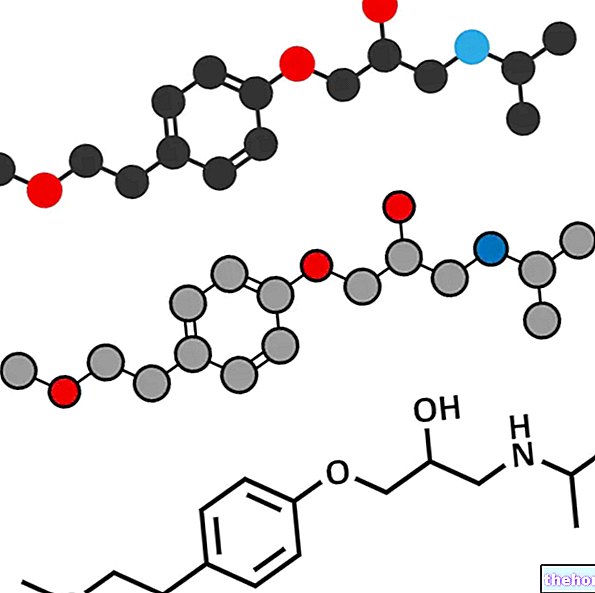
- Kaliumbesparende diuretika (givet for at reducere kaliumudskillelsen i urinen).
I de mest alvorlige tilfælde og / eller under stressende tilstande (indtræden af andre sygdomme, kirurgiske indgreb osv.) Kan påfyldning af kalium og andre mineralsalte udføres intravenøst, naturligvis skal en lignende operation udføres af sundhed specialiseret personale.