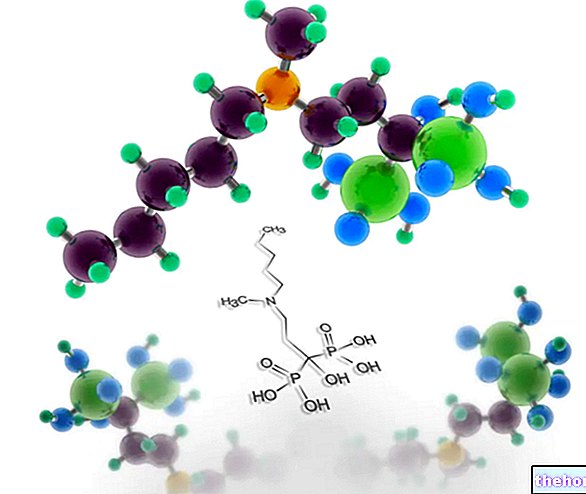
Aktive ingredienser: Irinotecan (Irinotecan hydrochloridtrihydrat)
Irinotecan Hospira 20 mg / ml koncentrat til infusionsvæske, opløsning
Hvorfor bruges Irinotecan - Generisk lægemiddel? Hvad er det for?
Irinotecan Hospira tilhører en gruppe lægemidler kaldet cytostatika (medicin mod kræft).
Irinotecan Hospira bruges til behandling af fremskreden metastatisk kræft i tyktarmen eller endetarmen hos voksne, og når sygdommen er på et fremskredent stadie i tarmen, enten i kombination med andre lægemidler mod kræft (kombinationsbehandling) eller alene (monoterapi).
Læger kan bruge en kombination af irinotecan med 5-fluorouracil / folinsyre (5-FU / FA) og bevacizumab til behandling af tyktarm og endetarmskræft.
Til behandling af tyktarm og endetarmskræft kan læger bruge en kombination af irinotecan med capecitabin med eller uden bevacizumab.
Til behandling af tyktarmskræft (KRAS vildtype), der udtrykker den epidermale vækstfaktorreceptor (EGFR), der er blokeret af monoklonalt antistof, kan din læge bruge kombinationen af irinotecan med cetuximab.
Spørg din læge for mere information om sygdommen.
Kontraindikationer Når Irinotecan - Generisk lægemiddel ikke bør anvendes
Brug ikke Irinotecan Hospira:
- Hvis du er følsom (allergisk) over for "Irinotecan hydrochlorid eller et af indholdsstofferne i dette lægemiddel (angivet i afsnit 6)
- Hvis du lider af andre tarmproblemer, eller hvis du har lidt af tarmobstruktion
- Hvis du ammer
- Hvis du har høje niveauer af bilirubin i dit blod (over tre gange den øvre grænse for normalområdet)
- Hvis du lider af alvorlig blodcellefejl (alvorlig knoglemarvsfejl)
- Hvis dit generelle helbred er dårligt (bestemt i henhold til internationale standarder)
- Hvis du bruger naturlægemidlet perikon (Hypericum perforatum)
For yderligere kontraindikationer til cetuximab eller bevacizumab eller capecitabin, som kan bruges i kombination med irinotecan, se oplysningerne om disse lægemidler.
Forholdsregler ved brug Hvad du skal vide, før du tager Irinotecan - Generisk lægemiddel
Denne medicin er kun beregnet til brug hos voksne. Kontakt lægen, hvis denne medicin er ordineret til brug hos et barn.
Der bør også udvises særlig forsigtighed hos ældre patienter.
Da Irinotecan Hospira er en kræftbekæmpende medicin, vil den blive givet til dig i en særlig afdeling og under opsyn af en læge, der er kvalificeret til brug af kræftbekæmpende lægemidler. Enhedens personale vil forklare dig, hvad du skal være omhyggelig med under og efter behandlingen. Denne indlægsseddel kan hjælpe dig med at huske dette.
Hvis du får irinotecan i kombination med cetuximab eller med bevacizumab eller capecitabin, skal du læse den indlægsseddel, der følger med pakningen med disse lægemidler.
Inden du bruger denne medicin, skal du fortælle det til din læge, hvis noget af følgende gælder for dig:
- Hvis du har hjerteproblemer.
- Hvis du ryger, lider du af forhøjet blodtryk eller højt kolesteroltal, da disse faktorer kan øge risikoen for hjerteproblemer under behandling med denne medicin.
- Hvis du har haft eller har brug for at blive vaccineret
Interaktioner Hvilke lægemidler eller fødevarer kan ændre virkningen af Irinotecan - Generic Drug
Fortæl det til din læge, hvis du tager eller for nylig har taget anden medicin, herunder receptpligtig medicin, dette gælder også for plantelægemidler.
Følgende medicin kan ændre virkningen af irinotecan:
- carbamazepin, phenobarbital eller phenytoin (lægemidler, der bruges til behandling af epilepsi)
- ketoconazol (bruges til behandling af svampeinfektioner)
- rifampicin (bruges til behandling af tuberkulose)
- Perikon (Hypericum perforatum) bør ikke tages under behandling med irinotecan eller mellem behandlingsforløb, da det kan reducere virkningen af irinotecan.
- Atazanavir (bruges til behandling af hiv)
- Antikoagulantia (bruges til at tynde blodet)
- Vacciner. Fortæl det til din læge, hvis du har haft eller skal have vaccinationer
- Ciclosporin eller tacromilus (bruges til at nedsætte immunsystemet)
Hvis du skal opereres, skal du fortælle det til din læge eller anæstesilæge, at du tager denne medicin, da det kan ændre virkningen af nogle lægemidler, der bruges under operationen.
Advarsler Det er vigtigt at vide, at:
Under administration af irinotecan (30 - 90 minutter) og op til 24 timer efter administration kan du opleve nogle af følgende symptomer:
- Diarré
- Svedende
- Mavesmerter
- Overdreven rive
- Synsforstyrrelser
- Overdreven spyt
Det medicinske udtryk for disse symptomer er "akut cholinerg syndrom", som kan behandles (med atropin). Hvis du oplever nogen af disse symptomer, skal du straks informere din læge, som vil give dig den mest egnede behandling.
Fra den første dag efter behandling med irinotecan og til den næste kan du opleve forskellige symptomer, som kan være alvorlige og kræve øjeblikkelig behandling og omhyggeligt tilsyn. Disse symptomer er:
Diarré
Hvis diarré starter mere end 24 timer efter irinotecan -behandling ("forsinket diarré"), kan det være alvorligt. Det sker ofte omkring 5 dage efter behandlingen. Denne diarré skal behandles straks og overvåges nøje. Følg nedenstående instruktioner umiddelbart efter den første diarréudledning:
- Tag den behandling mod diarré, som din læge har givet dig, følg nøjagtigt de modtagne instruktioner. Behandlingen bør ikke ændres uden først at tjekke med din læge. Den anbefalede behandling mod diarre er loperamid (4 mg første gang og derefter 2 mg hver 2. time, selv om natten). Dette bør fortsættes i mindst 12 timer efter den sidste diarréudledning.Den anbefalede dosis for loperamid bør ikke tages i mere end 48 timer.
- Drik straks store mængder vand, fugtgivende væsker (f.eks. Vand, sodavand, bouillon eller oral fugtgivende behandling).
- Informer straks den læge, der fører tilsyn med behandlingen af diarré. Hvis du ikke kan nå det, skal du kontakte hospitalet og operationsenheden, der fører tilsyn med behandlingen med irinotecan. Det er meget vigtigt, at de er informeret om diarré.
Til behandling af diarré anbefales hospitalsindlæggelse i følgende tilfælde:
- Du lider af diarré og har feber (over 38 ° C)
- Du har alvorlig diarré (og opkastning) med stort vandtab, der kræver intravenøs hydrering
- Du lider stadig af diarré 48 timer efter påbegyndelse af behandlingen mod diarre
Bemærk! Følg ikke andre behandlinger og væskeindtag for diarré end dem, der er ordineret af din læge. Følg din læge instruktioner. Selvom du har oplevet forsinket diarré i tidligere cyklusser, bør anti-diarrébehandling ikke bruges til forebyggelse.
Feber
Hvis din kropstemperatur stiger over 38 ° C, kan det være tegn på infektion, især hvis du også har diarré. Hvis du har feber (højere end 38 ° C), skal du straks kontakte din læge eller hospitalet for at starte den nødvendige behandling.
Kvalme (opkastning) og opkastning
Hvis du lider af kvalme og / eller opkastning, skal du straks kontakte din læge eller hospitalet.
Neutropeni
Irinotecan kan forårsage et fald i antallet af nogle hvide blodlegemer, der spiller en vigtig rolle i bekæmpelsen af infektioner. Dette er kendt som neutropeni. Neutropeni rapporteres ofte efter irinotecan -behandling og er reversibel. Din læge skal regelmæssigt foretage blodprøver for at overvåge disse hvide blodlegemer. Neutropeni er alvorlig og skal behandles hurtigt og nøje overvåges.
Besvær med at trække vejret
Hvis du har svært ved at trække vejret, skal du straks kontakte din læge.
Nedsat leverfunktion
Inden behandlingen påbegyndes med irinotecan og før hvert behandlingsforløb, tjekker din læge din leverfunktion (ved hjælp af blodprøver).
Nedsat nyrefunktion
Denne medicin er ikke testet hos patienter med nyreproblemer. Hvis du har nyreproblemer, skal du kontakte din læge.
Efter hjemkomsten fra hospitalet, hvis du får et eller flere af ovenstående symptomer, skal du straks kontakte din læge eller hospitalsenheden, der fører tilsyn med din behandling med irinotecan.
Graviditet og amning
Brug ikke Irinotecan Hospira:
- Hvis du ammer
Du må ikke behandles med irinotecan, hvis du er gravid, medmindre den kliniske tilstand kræver behandling med irinotecan.
Hvis du eller din partner behandles med irinotecan, skal du undgå at blive gravid under behandlingen. Kvinder i den fertile alder og mænd skal anvende passende præventionsmetoder under behandlingen og i mindst:
- Hos kvinder en måned efter behandlingens afslutning
eller
- Hos mænd tre måneder efter behandlingens afslutning
Hvis du også bliver gravid i løbet af denne periode, skal du straks informere din læge.
Kørsel og brug af maskiner
I nogle tilfælde kan Irinotecan Hospira forårsage bivirkninger, der påvirker evnen til at føre motorkøretøj og betjene maskiner. Hvis du er i tvivl, skal du kontakte din læge eller apotek.
Du kan opleve svimmelhed eller synsforstyrrelser i løbet af de første 24 timer efter behandling med Irinotecan Hospira. Skulle dette ske for dig, må du hverken køre bil eller bruge maskiner.
Irinotecan Hospira indeholder sorbitol. Hvis din læge har fortalt dig, at du ikke tåler nogle sukkerarter (f.eks. Fructoseintolerance), skal du kontakte din læge, inden du tager dette lægemiddel. Dette lægemiddel indeholder mindre end 1 mmol natrium (23 mg) pr. Dosis, dvs. i det væsentlige "natriumfrit".
Dosering og anvendelsesmåde Sådan bruges Irinotecan - Generisk lægemiddel: Dosering
Tag altid dette lægemiddel nøjagtigt efter lægens eller apotekspersonalets anvisning. Hvis du er i tvivl, skal du kontakte din læge eller apotek.
Kun for voksne.
Irinotecan vil blive givet som infusion i en vene i en periode på 30-90 minutter.
Den dosis, der skal infunderes, afhænger af din alder, højde, vægt og din helbredstilstand.Lægen vil beregne dit kropsoverfladeareal i kvadratmeter (m2) baseret på din højde og vægt. Doseringen afhænger også af andre behandlinger, du måtte have modtaget for din kræftsygdom.
- Hvis du allerede er blevet behandlet med 5-fluorouracil, vil du normalt blive behandlet med irinotecan alene med en dosis på 350 mg / m2 hver tredje uge.
- Hvis du aldrig er blevet behandlet med kemoterapi, modtager du normalt 180 mg / m2 irinotecan hver anden uge efterfulgt af folinsyre og 5-fluorouracil.
Hvis du behandles med irinotecan i kombination med cetuximab, kan irinotecan ikke administreres i en time efter afslutningen af cetuximab -infusionen.
Følg altid din læges råd vedrørende løbende behandling.
Disse doser kan justeres af din læge i henhold til din tilstand og eventuelle uønskede virkninger, du måtte opleve.
Overdosering Hvad skal man gøre, hvis man har taget for meget Irinotecan - Generisk lægemiddel
Hvis du får en højere dosis irinotecan end nødvendigt, kan bivirkningerne være mere alvorlige. Du får maksimal støtte til at forhindre dehydrering sekundært til diarré og til behandling af infektiøse komplikationer. Hvis du mener, at du er blevet behandlet med en højere dosis, skal du kontakte din læge.
Hvis du har yderligere spørgsmål til brugen af denne medicin, skal du tale med din læge eller apotek eller sygeplejerske.
Bivirkninger Hvad er bivirkningerne af Irinotecan - Generic Drug
Ligesom al anden medicin kan denne medicin forårsage bivirkninger, men ikke alle får bivirkninger.
Din læge vil informere dig om disse bivirkninger og forklare risici og fordele ved behandlingen.
Nogle af disse bivirkninger skal behandles med det samme, disse er:
- diarré
- et fald i antallet af neutrofile granulocytter, en type hvide blodlegemer, som spiller en vigtig rolle i bekæmpelsen af infektioner
- feber
- kvalme og opkast
- åndedrætsbesvær (sandsynligvis symptom på alvorlige allergiske reaktioner)
Læs instruktionerne i afsnittet "Advarsler og forholdsregler" omhyggeligt, og følg dem, hvis du oplever nogen af de effekter, der er beskrevet ovenfor.
Andre bivirkninger omfatter:
Meget almindelige bivirkninger (hos mere end 1 ud af 10 patienter)
- blodsygdomme, herunder et unormalt lavt antal neutrofile granulocytter, en type hvide blodlegemer (neutropeni) og et fald i mængden af hæmoglobin i blodet (anæmi)
- i kombinationsbehandling, trombocytopeni (reduktion i antallet af blodplader) med resulterende blå mærker, tendens til blødning og unormal blødning
- alene, feber
- ved monoterapi, infektioner
- alvorlig forsinket diarré
- alene, alvorlig kvalme (opkastning) og opkastning (kvalme)
- hårtab (hår vokser tilbage, når behandlingen er afsluttet)
- i kombinationsbehandling, forbigående mild til moderat stigning i serumniveauer af visse leverenzymer (SGPT, SGOT, alkalisk phosphatase) eller bilirubin
Almindelige bivirkninger (hos færre end 1 ud af 10 patienter, men mere end 1 ud af 100)
- alvorligt akut forbigående kolinergt syndrom: De vigtigste symptomer er defineret som tidlig diarré og flere andre symptomer såsom mavesmerter; røde, smertefulde, kløende eller vandige øjne (konjunktivitis) løbende næse (rhinitis); lavt blodtryk; rødme sekundært til dilatation af blodkar (vasodilatation); svedtendens; kuldegysninger; en følelse af generel utilpashed og sygdom svimmelhed synsforstyrrelser; sammentrækning af eleven; revner og øget spytdannelse, der forekommer under eller inden for de første 24 timer efter infusion af Irintoecan Hospira
- ved monoterapi, trombocytopeni (reduktion i antallet af blodplader), der forårsager blå mærker, tendens til blødning og unormal blødning
- i kombinationsbehandling, feber
- i kombinationsbehandling, infektioner
- infektioner forbundet med et alvorligt fald i antallet af visse typer hvide blodlegemer (neutropeni), hvilket resulterede i 3 dødsfald
- feber forbundet med et alvorligt fald i antallet af nogle hvide blodlegemer (febril neutropeni)
- i kombinationsbehandling, alvorlig kvalme (opkastning) og opkastning (kvalme)
- vandtab (dehydrering), ofte forbundet med diarré og / eller opkastning
- forstoppelse
- føler sig svag (asteni)
- ved monoterapi, forbigående mild til moderat stigning i serumniveauer af nogle leverenzymer (transaminase, alkalisk phosphatase) eller bilirubin
- forbigående let til moderat stigning i kreatininniveauer i blodet
- i kombinationsbehandling, forbigående og markant (grad 3) stigning i serumbilirubinniveauer
Ikke almindelige bivirkninger (hos færre end 1 ud af 100 patienter, men mere end 1 ud af 1.000)
- milde allergiske reaktioner (hudrødme inklusive kløende rød hud, nældefeber, konjunktivitis, rhinitis)
- milde hudreaktioner
- på infusionsstedet, beskedne reaktioner
- lungesygdom, der manifesterer sig som åndenød, tør hoste og inspirerende rale (interstitiel lungesygdom); tidlige virkninger såsom vejrtrækningsbesvær
- delvis eller total tarmobstruktion (tarmobstruktion, tarmblokering), mave og blødning af tarmen
- tarmbetændelse, der forårsager mavesmerter og / eller diarré (en tilstand kendt som pseudomembranøs colitis)
- nyresvigt, lavt blodtryk eller hjertedekompensation hos patienter med tidligere episoder med dehydrering forbundet med diarré og / eller opkastning eller sepsis
Sjældne bivirkninger (hos færre end 1 ud af 1.000 patienter, men mere end 1 ud af 10.000)
- alvorlige allergiske reaktioner (anafylaktisk / anafylaktoid reaktion), som omfatter hævelse af hænder, fødder, ankler, ansigt, læber, mund eller hals, som kan forårsage synkebesvær eller ekstremt svært vejrtrækning
- muskeltrækninger eller kramper eller følelsesløshed (paræstesi)
- betændelse i tyktarmen med resulterende mavesmerter (colitis inklusive tyfitis, iskæmisk og ulcerøs colitis)
- tarmperforering
- mistet appetiten
- mavesmerter
- betændelse i slimhinderne
- reducerede niveauer af kalium og natrium i blodet, hovedsageligt relateret til diarré og opkastning
- symptomatisk og asymptomatisk betændelse i bugspytkirtlen (især mavesmerter)
- forhøjelse af blodtrykket under og efter behandlingen
Meget sjældne bivirkninger (hos færre end 1 ud af 10.000 patienter)
- reversible taleproblemer
- øgede niveauer af nogle fordøjelsesenzymer, der metaboliserer sukker (amylaser) og fedtstoffer (lipaser)
- et tilfælde med lavt blodpladetal sekundært tilstedeværelsen af antistoffer mod blodplader
Bivirkninger med hyppighed ikke kendt:
- udslæt
- unormalt lavt antal hvide blodlegemer (leukopeni).
Hvis du behandles med irinotecan i kombination med cetuximab, kan nogle af de bivirkninger, du oplever, være sekundære i forhold til denne kombination. Sådanne bivirkninger kan omfatte acne-lignende rødme. Derfor skal du også læse indlægssedlen, der fulgte med cetuximab -pakken.
Hvis du behandles med irinotecan i kombination med capecitabin, kan nogle af de bivirkninger, du oplever, være sekundære i forhold til denne kombination. Disse bivirkninger kan omfatte: meget almindelige blodpropper, almindelige allergiske reaktioner, hjerteanfald og feber hos patienter med lavt antal hvide blodlegemer. Derfor skal du også læse indlægssedlen, der fulgte med capecitabinpakningen.
Hvis du behandles med irinotecan i kombination med capecitabin og bevacizumab, kan nogle af de bivirkninger, du oplever, være sekundære i forhold til denne kombination. Sådanne bivirkninger kan omfatte: reduceret antal hvide blodlegemer, blodpropper, forhøjet blodtryk og hjerteanfald. Derfor skal du også læse indlægssedlerne for capecitabin og bevacizumab.
Indberetning af bivirkninger
Tal med din læge eller apotek, hvis du får bivirkninger, herunder mulige bivirkninger, der ikke er anført i denne indlægsseddel. Uønskede virkninger kan også rapporteres direkte via det nationale rapporteringssystem på http://www.agenziafarmaco.gov.it/it/responsabili. Ved at rapportere bivirkninger kan du hjælpe med at give mere information om lægemidlets sikkerhed..
Udløb og opbevaring
Sådan opbevares Irinotecan Hospira
- Opbevar denne medicin utilgængeligt for børn.
- Brug ikke dette lægemiddel efter den udløbsdato, der står på den ydre karton og hætteglassets etiket. Udløbsdatoen refererer til den sidste dag i måneden.
- Koncentrat: For at beskytte medicinen mod lys, skal hætteglasset opbevares i den originale emballage. Må ikke fryses. Efter første åbning, da de ikke indeholder et antimikrobielt konserveringsmiddel, skal hætteglassene bruges med det samme.
- Fortyndet koncentrat: Kun til engangsbrug. Restopløsningen skal kasseres.
- Efter fortynding: Kemisk og fysisk stabilitet i brug er påvist i glucose 50 mg / ml (5%) og natriumchlorid 9 mg / ml (0,9%) i 72 timer mellem 2 ° C og 8 ° C. Fra et mikrobiologisk synspunkt bør medicinen bruges straks. Hvis den ikke bruges med det samme, forbliver opbevaringstider og -betingelser før brug brugerens ansvar og vil normalt ikke overstige 24 timer ved 2 ° C til 8 ° C, medmindre fortynding har fundet sted. Under kontrollerede og validerede aseptiske forhold.
Brug ikke denne medicin, hvis du bemærker synlige partikler i koncentratet eller infusionsopløsningen.
Bortskaf ikke medicin via spildevand eller husholdningsaffald. Spørg din apotek om, hvordan du skal bortskaffe medicin, der ikke længere er anvendelige. Disse foranstaltninger hjælper med at beskytte miljøet.
Deadline "> Andre oplysninger
Irinotecan Hospira indeholder
- Det aktive stof er irinotecanhydrochloridtrihydrat. Hver milliliter (ml) opløsning indeholder 20 milligram (mg) irinotecanhydrochloridtrihydrat svarende til 17,33 mg irinotecan.
- Øvrige indholdsstoffer er sorbitol (E420), mælkesyre og vand til injektionsvæsker og natriumhydroxid, saltsyre (for at korrigere pH).
Irinotecanhydrochlorids udseende og pakningens indhold
Irinotecan Hospira findes i den farmaceutiske form af koncentrat til infusionsvæske, opløsning (en koncentreret opløsning, der fortyndes, før den administreres ved langsom intravenøs infusion).
Denne medicin er pakket i hætteglas med 2 ml, 5 ml og 25 ml irinotecanhydrochloridtrihydrat.
Hætteglassene er dækket med et beskyttende plastlag, der reducerer risikoen for spild, hvis hætteglasset går i stykker - disse kendes under betegnelsen ONCO -TAIN. Hætteglassene fås i enkeltpakninger.
Ikke alle pakningsstørrelser er nødvendigvis markedsført.
Deadline "> Information til sundhedspersonale
Følgende oplysninger er kun beregnet til sundhedspersonale
BRUGSANVISNING, HÅNDTERING OG BORTSKAFFELSE
Som med andre potentielt giftige forbindelser skal der udvises forsigtighed ved håndtering eller fremstilling af irinotecan -opløsninger.
Instruktioner om brug / håndtering
Som med andre antineoplastiske lægemidler skal irinotecan tilberedes og håndteres forsigtigt. Brug af beskyttelsesbriller, maske og handsker er påkrævet. Gravide kvinder må aldrig håndtere cytotoksiske midler. Hvis opløsningen af irinotecankoncentrat til infusion eller opløsningen, der er forberedt til infusionen, skal komme i kontakt med huden, skal den vaskes straks og rigeligt med sæbe og vand. Hvis irinotecan koncentrat til infusion eller infusionsvæsken skal komme i kontakt med slimhinder, vaskes straks med vand.
Forberedelse af den intravenøse infusion
Som med alle andre injicerbare lægemidler skal Irinotecan -opløsningen tilberedes aseptisk.
Hvis der observeres et bundfald i hætteglassene eller infusionsvæsken, skal produktet kasseres efter standard hospitalsprocedurer, der gælder for cytotoksiske lægemidler.
Under aseptiske forhold trækkes den nødvendige mængde af den koncentrerede Irinotecan -opløsning op fra hætteglasset ved hjælp af en gradueret sprøjte og injiceres i en 250 ml infusionspose eller flaske, der kun indeholder natriumchlorid 9 mg / ml (0,9%) eller glucoseopløsning 50 mg / ml ( 5%). Infusionen skal derefter blandes perfekt ved manuel rotation.
Eliminering
Alt materiale, der bruges til fortynding og administration, skal kasseres i henhold til standard hospitalsprocedurer, der gælder for cytotoksiske lægemidler.
Indlægsseddel: AIFA (Italian Medicines Agency). Indhold offentliggjort i januar 2016. De foreliggende oplysninger er muligvis ikke opdaterede.
For at få adgang til den mest opdaterede version er det tilrådeligt at få adgang til webstedet AIFA (Italian Medicines Agency). Ansvarsfraskrivelse og nyttige oplysninger.
01.0 LÆGEMIDLETS NAVN -
IRINOTECAN HOSPIRA 20 MG / ML KONCENTRAT TIL LØSNING TIL INFUSION
02.0 KVALITATIV OG KVANTITATIV SAMMENSÆTNING -
En milliliter indeholder 20 mg Irinotecan hydrochloridtrihydrat, svarende til 17,33 mg Irinotecan.
Et hætteglas med 2 ml injektionsvæske, opløsning indeholder 40 mg Irinotecan hydrochloridtrihydrat
Et hætteglas med 5 ml injektionsvæske, opløsning indeholder 100 mg Irinotecan hydrochloridtrihydrat
Et hætteglas med 25 ml injektionsvæske, opløsning indeholder 500 mg Irinotecan hydrochloridtrihydrat
Hjælpestoffer:
Indeholder Sorbitol, (E420) 45,0 mg / ml
Den fulde liste over hjælpestoffer findes i afsnit 6.1
03.0 LÆGEMIDDELFORM -
Koncentrat til infusionsvæske, opløsning.
En klar, farveløs til lysegul opløsning.
04.0 KLINISKE OPLYSNINGER -
04.1 Terapeutiske indikationer -
Irinotecan er indiceret til behandling af avanceret tyktarmskræft.
• I kombination med 5-fluorouracil (5-FU) og folinsyre (FA) hos patienter, der ikke tidligere er blevet behandlet med kemoterapi for fremskreden sygdom
• Som monoterapi hos patienter, hvor konventionel 5-fluorouracil-holdig behandling har mislykkedes
Irinotecan i kombination med Cetuximab er indiceret til behandling af patienter med vildtype KRAS-udtrykkende epidermal vækstfaktor (EGFR) receptor-udtrykkende metastatisk kolorektal cancer, som ikke tidligere er blevet behandlet for metastatisk sygdom eller efter svigt i cytotoksisk behandling indeholdende irinotecan ( se afsnit 5.1).
Irinotecan i kombination med 5-FU, FA og Bevacizumab er indiceret ved førstelinjebehandling af patienter med metastatisk kræft i tyktarmen eller endetarmen.
04.2 Dosering og indgivelsesmåde -
Kun til brug hos voksne patienter. Den fortyndede Irinotecan infusionsvæske, opløsning, skal infunderes i en central eller perifer vene.
Anbefalet dosering
Doseringerne af Irinotecan, der er angivet i denne produktresumé, refererer til mg Irinotecan -hydrochloridtrihydrat.
Som monoterapi (for tidligere behandlede patienter)
Den anbefalede dosis Irinotecan er 350 mg / m² administreret ved intravenøs infusion, der varer 30 - 90 minutter hver 3. uge (se pkt. 4.4 og 6.6).
I kombinationsbehandling (til tidligere ubehandlede patienter)
Sikkerheden og effekten af Irinotecan i kombination med 5-fluorouracil (5FU) og folinsyre (FA) blev bestemt med følgende behandlingsregime (se pkt.5.1)
• Irinotecan hydrochlorid plus 5-FU / AF hver 2. uge
Den anbefalede dosis Irinotecan er 180 mg / m² administreret hver anden uge som en intravenøs infusion, der varer 30-90 minutter, efterfulgt af infusion af FA og 5 -FU.
For information om dosering og indgivelsesmåde samtidig med Cetuximab, se oplysningerne om dette lægemiddel. Normalt anvendes den samme dosis Irinotecan som den, der blev givet i den sidste cyklus af det tidligere Irinotecan-holdige regime.Irinotecan bør ikke administreres tidligere end en time efter afslutningen af Cetuximab-infusionen.
For dosering og indgivelsesmåde for Bevacizumab henvises til det respektive produktresumé.
Dosisjustering
Irinotecan bør administreres, efter at alle NCI-CTC (National Cancer Institute Common Toxicity Criteria) grad 0 eller 1 bivirkninger er forsvundet, og behandlingsrelateret diarré er fuldstændig forsvundet.
Ved begyndelsen af et efterfølgende infusionsforløb bør dosis af Irinotecan og eventuelt 5-FU reduceres i overensstemmelse med graden af mere alvorlige bivirkninger observeret under den foregående infusion. Behandlingen bør udsættes i 1-2 uger, indtil fuldstændig helbredelse fra behandlingsrelaterede bivirkninger.
Med følgende bivirkninger skal du anvende en 15-20% dosisreduktion af Irinotecan og / eller 5-FU efter behov:
• hæmatologisk toksicitet (neutropeni grad 4), febril neutropeni (neutropeni grad 3-4 og feber grad 2-4), trombocytopeni og leukopeni (grad 4),
• ikke-hæmatologisk toksicitet (grad 3-4).
Anbefalinger til dosisjustering af Cetuximab, når det gives i kombination med Irinotecan, bør følges i henhold til produktinformationen for dette specifikke lægemiddel.
For bevacizumab dosisjustering, når det gives i kombination med Irinotecan / 5-FU / FA, se bevacizumabs resumé af produktkarakteristika.
Behandlingens varighed
Irinotecan -behandlingen bør fortsættes, så længe der observeres objektiv progression af sygdommen, og der ikke forekommer symptomer på uacceptabel toksicitet.
Særlige populationer
Patienter med nedsat leverfunktion:
Monoterapi
Bilirubinværdier (op til 3 gange den øvre grænse for det normale område), hos patienter med WHO's præstationsstatus ≤2, bestemmer den initiale dosis af Irinotecan.For disse patienter med hyperbilirubinæmi og en protrombintid større end 50%, clearance af Irinotecan reduceres (se pkt. 5.2), derfor er der en øget risiko for hæmatisk toksicitet.Derfor bør ugentlige fuldstændige blodtællinger overvåges i denne patientpopulation.
• Hos patienter med bilirubinværdier op til 1,5 gange den øvre grænse for det normale område er den anbefalede dosis Irinotecan 350 mg / m².
• Hos patienter med bilirubinværdier mellem 1,5 - 3 gange den øvre grænse for det normale område er den anbefalede dosis Irinotecan 200 mg / m².
• Patienter med bilirubinværdier større end 3 gange den øvre grænse for det normale område bør ikke behandles med Irinotecan (se pkt. 4.3 og 4.4).
Der er ingen data tilgængelige for patienter med nedsat leverfunktion behandlet med irinotecan i kombinationsbehandling.
Patienter med nedsat nyrefunktion
Da der ikke er udført specifikke undersøgelser med dette lægemiddel i denne patientgruppe, anbefales brug af Irinotecan ikke til patienter med nedsat nyrefunktion (se pkt. 4.4 og 5.2).
Ældre borgere
Der er ikke udført specifikke farmakokinetiske undersøgelser hos ældre. På grund af den øgede hyppighed af nedsatte vitale funktioner, bør valg af dosis foretages med forsigtighed i denne population. Disse patienter kræver mere overvågning (se pkt. 4.4).
04.3 Kontraindikationer -
Kronisk inflammatorisk tarmsygdom og / eller tarmobstruktion (se pkt. 4.4).
Tidligere alvorlige overfølsomhedsreaktioner over for Irinotecan hydrochloridtrihydrat eller over for et eller flere af hjælpestofferne i Irinotecan.
Amning (se afsnit 4.6 og 4.4).
Bilirubinværdier> 3 gange den øvre grænse for normalområdet (se pkt. 4.4).
Alvorlig knoglemarvsfejl.
"WHO's præstationsstatus"> 2.
Samtidig brug af perikon (se afsnit 4.5).
For yderligere kontraindikationer af Cetuximab eller Bevacizumab, se produktinformationen for disse lægemidler.
04.4 Særlige advarsler og passende forholdsregler ved brug -
Anvendelse af Irinotecan bør forbeholdes enheder specialiseret i administration af cytotoksiske terapier under opsyn af en læge, der er kvalificeret til brug af antineoplastiske behandlinger.
I betragtning af bivirkningernes art og forekomst, bør Irinotecan kun ordineres i følgende tilfælde efter vurdering af den forventede fordel i forhold til eventuelle risikofaktorer:
• patienter med risikofaktorer, især dem med en WHO = 2 "Performance Status".
• i de sjældne tilfælde, hvor patientens tilslutning til instruktionerne til behandling af bivirkninger er forudsigelig (behov for øjeblikkelig og langvarig antidiarrheal behandling forbundet med indtagelse af store mængder væske ved begyndelsen af forsinket diarré). en "omhyggelig overvågning på hospitalet.
Når Irinotecan bruges alene, gives det normalt i henhold til doseringsplanen hver 3. uge. Ugeplanen (se afsnit 5) kan dog overvejes for patienter, der har brug for hyppigere overvågning, eller som er særligt udsatte for alvorlig neutropeni.
Forsinket diarré
Patienter bør være opmærksom på risikoen for forsinket diarré, som kan forekomme mere end 24 timer efter administration af Irinotecan og til enhver tid forud for den næste cyklus. Ved monoterapi var den gennemsnitlige tid til første væskeevakuering dag 5. efter infusion af Irinotecan. Patienter bør straks underrette deres læge, hvis der opstår diarré og starte passende behandling med det samme.
Patienter med øget risiko for diarré er dem, der tidligere blev behandlet med abdominal / bækkenstrålebehandling, patienter med basal hyperleukocytose og patienter med "Performance Status"> 2 og kvinder. Hvis den ikke behandles korrekt, kan diarré true overlevelse, især i tilfælde hvor patienten samtidig er neutropen.
Så snart den første flydende afføring dukker op, skal patienten begynde at drikke store mængder væsker i form af drikkevarer, der indeholder elektrolyt, og passende antidiarrheal terapi startes med det samme. Passende antidiarrheal behandling vil blive ordineret af den læge, der administrerede Irinotecan. Efter udskrivning fra hospitalet skal patienter have den foreskrevne medicin til rådighed, så de kan behandle diarré, så snart den opstår.De bør også informere deres læge eller den afdeling, der administrerede Irinotecan om begyndelsen af diarré.
Den aktuelt anbefalede antidiarrheal-behandling er højdosis Loperamid (4 mg til at begynde med og derefter 2 mg hver 2. time). Denne behandling bør fortsætte i 12 timer efter den sidste flydende afføring og bør ikke ændres.Loperamid må under ingen omstændigheder administreres ved disse doser i mere end 48 timer på grund af risikoen for paralytisk ileus, og behandlingen bør vare mindst 12 timer .
Når diarré er forbundet med alvorlig neutropeni (neutrofiltal / mm³), bør bredspektret antibiotikaprofylakse tilføjes til antidiarrheal -behandlingen.
Ud over antibiotikabehandling anbefales hospitalsindlæggelse til kontrol af diarré i følgende tilfælde:
§ Diarré forbundet med feber,
§ Alvorlig diarré (f.eks. Behov for intravenøs rehydrering),
§ Vedvarende diarré 48 timer efter behandlingsstart med høje doser Loperamid.
Loperamid bør ikke gives som profylakse, heller ikke hos patienter, der har oplevet forsinket diarré i tidligere behandlingsforløb med lægemidlet.
Dosisreduktion anbefales til patienter, der har oplevet alvorlig diarré i efterfølgende cyklusser (se 4.2).
Hæmatologi
Det anbefales at foretage en komplet ugentlig kontrol af blodtællinger under behandling med Irinotecan. Patienterne bør være opmærksom på risikoen for neutropeni og betydningen af feber. Febral neutropeni (temperatur> 38 ° C og neutrofiltal ≤ 1000 celler / mm³) skal behandles akut på hospitalet med intravenøst bredspektret antibiotika.
Hos patienter, der har oplevet alvorlige hæmatologiske hændelser, anbefales en dosisreduktion ved efterfølgende administration (se pkt.4.2).
Risikoen for infektioner og hæmatologisk toksicitet øges hos patienter med alvorlig diarré. Et fuldstændigt blodtal bør kontrolleres hos disse patienter.
Patienter med nedsat aktivitet af uridindiphosphatglucuronosyltransferase (UGT1A1)
SN-38 afgiftes fra UGT1A1 til SN-38 glucuronid. Personer med genetisk mangel på UGT1A1 (Crigler-Najjar syndrom type 1 og type 2 eller personer, der er homozygote for UGT1A1 * 28-allelen [Gilberts syndrom]) har en øget risiko for Irinotecan-toksicitet. Overvej at bruge en reduceret startdosis Irinotecan.
Ændret leverfunktion
Leverfunktionstest bør udføres under baseline betingelser og før hver behandlingscyklus.
Ugentlig overvågning af komplette blodlegemer bør udføres hos patienter med bilirubinværdier 1,5 til 3 gange den øvre grænse for normalområdet på grund af nedsat clearance af Irinotecan (se pkt. 5.2), og derfor udgør disse en højere risiko for hæmatotoksicitet. For patienter med bilirubinværdier> 3 gange den øvre grænse for normalområdet, se afsnit 4.3.
Kvalme og opkast
Profylaktisk behandling med et antiemetikum anbefales før hver behandling med Irinotecan. Kvalme og opkastning er ofte blevet rapporteret. Patienter med opkastning forbundet med forsinket diarré bør indlægges hurtigst muligt.
Akut kolinergt syndrom
Hvis akut cholinergt syndrom (defineret som tidlig diarré forbundet med forskellige andre tegn og symptomer såsom svedtendens, mavekramper, miose og spyt) forekommer, bør atropinsulfat (0,25 mg subkutant) gives, medmindre det er til stede. Kliniske kontraindikationer (se pkt.4.8).
Der skal udvises forsigtighed hos patienter med astma. Hvis patienten rapporterer om akut og alvorligt cholinergt syndrom, anbefales den profylaktiske anvendelse af atropinsulfat ved efterfølgende administration af Irinotecan.
Respiratoriske sygdomme
Interstitielle lungesygdomme, der optræder som lungeinfiltrater, er ualmindelige under irinotecan -behandling. Interstitiel lungesygdom kan være dødelig. De sandsynlige risikofaktorer forbundet med udviklingen af lungeinfiltrater er brug af lægemidler, der er giftige for lungerne, strålebehandling og brug af vækstfaktorer. Patienter med risikofaktorer bør overvåges nøje for luftvejssymptomer før og under behandling med Irinotecan.
Ekstravasation
Selvom irinotecan ikke blærer, skal der udvises forsigtighed for at undgå ekstravasation, og infusionsstedet skal observeres for tegn på betændelse.I tilfælde af ekstravasation, rødme på injektionsstedet, vask af stedet anbefales. Og påføring af is.
Hjertepatologier
Myokardielle iskæmiske hændelser er blevet observeret efter irinotecan-behandling, overvejende hos patienter med allerede eksisterende hjertesygdom, med andre kendte risikofaktorer for hjertesygdomme eller med en historie med cytotoksisk kemoterapi (se pkt. 4.8 Bivirkninger).
Som følge heraf skal patienter med kendte risikofaktorer overvåges nøje, og der skal træffes passende foranstaltninger for at minimere variable risikofaktorer (f.eks. Rygning, hypertension og hyperlipidæmi)
Immunsuppressive virkninger / øget modtagelighed for infektioner Administration af levende eller svækkede vacciner til patienter immunkompromitteret af kemoterapeutiske midler, herunder irinotecan, kan forårsage alvorlige eller dødelige infektioner. Vaccination med en levende vaccine bør undgås hos patienter behandlet med irinotecan. De kan administreres. inaktiveret; responsen på sådanne vacciner kan dog reduceres.
Ældre borgere
Hos ældre patienter, på grund af den højere hyppighed af nedsatte biologiske funktioner, f.eks. Leverfunktion, kræver dosisreduktion af Irinotecan i denne population større forsigtighed (se pkt.4.2).
Patienter med tarmobstruktion
Patienter bør ikke behandles med Irinotecan, før tarmobstruktionen er løst (se pkt. 4.3).
Patienter med nedsat nyrefunktion
Der er ikke udført specifikke undersøgelser af denne population (se pkt. 4.2 og 5.2).
Andre
Lægemidlet er uegnet til patienter med arvelig fructoseintolerance, da det indeholder sorbitol. Sjældne tilfælde af nyresvigt, hypotension eller kredsløbssvigt er blevet observeret hos patienter med dehydrering forbundet med diarré og / eller opkastning eller med sepsis.
Brug af prævention er påkrævet under og i mindst 3 måneder efter behandlingens afslutning hos kvinder i den fertile alder og hos mænd (se pkt. 4.6).
Samtidig behandling af Irinotecan med en stærk hæmmer (f.eks. Ketoconazol) eller inducer (f.eks. Rifampicin, Carbamazepin, Phenobarbital, Phenytoin, perikon) af Cytochrome P4503A4 (CYP3A4) kan ændre Irinotecans metabolisme og bør undgås. ).
04.5 Interaktioner med andre lægemidler og andre former for interaktion -
Interaktioner mellem Irinotecan og neuromuskulært blokerende lægemidler kan ikke udelukkes. Da Irinotecan har anticholinesterase-aktivitet, kan lægemidler med anticholinesterase-aktivitet forlænge den neuromuskulære blokerende virkning af suxamethonium og modvirke neuromuskulær blokade fra ikke-depolariserende lægemidler.
Talrige undersøgelser har vist, at samtidig behandling med cytokrom P450 3A (CYP3A) -inducerende antikonvulsiva (f.eks. Carbamazepin, Phenobarbital eller Phenytoin) resulterer i reduceret eksponering for Irinotecan, SN-38 og SN-38 glucuronat og reduceret effekt farmakodynamik.
Virkningerne af disse antikonvulsive lægemidler afspejlede sig i et fald i SN-38 og SN-38G AUC med 50% eller mere.Ud over induktion af P450 3A-enzymer kan øget glukuronidering og øget galdeudskillelse spille en rolle.Vigtig ved reduceret eksponering til Irinotecan og dets metabolitter.
En undersøgelse viste, at samtidig behandling med ketoconazol resulterede i et fald i AUC for dets største oxidative metabolit APC med 87% og en stigning i AUC for SN-38 med 109% i forhold til irinotecan administreret alene. Der skal udvises forsigtighed. på samtidig behandling med lægemidler, der vides at hæmme (f.eks. Ketoconazol) eller fremkalde (f.eks. Carbamazepin, Phenobarbital, Phenytoin, Rifampicin) lægemiddelmetabolisme via P450 3A4. Samtidig behandling af Irinotecan med en hæmmer / inducer af denne rutemetabolisme kan ændre metabolismen af irinotecan og bør undgås (se pkt. 4.4).
I et lille farmakokinetisk studie (n = 5), hvor Irinotecan 350 mg / m² blev administreret samtidigt med 900 mg perikon (Hypericum perforatum), blev der rapporteret et fald på 42% i plasmakoncentrationer af den aktive metabolit af Irinotecan, SN-38.
Perikon reducerer plasmaniveauerne af SN-38, derfor bør det ikke administreres sammen med Irinotecan (se pkt. 4.3).
Kombineret behandling af 5-FU / FA ændrer ikke Irinotecans farmakokinetik.
Der er ingen tegn på, at sikkerhedsprofilen for Irinotecan påvirkes af Cetuximab eller omvendt.
I en undersøgelse var koncentrationer af Irinotecan ens hos patienter behandlet med Irinotecan / 5-FU / FA alene eller i kombination med Bevacizumab. Koncentrationer af SN-38, den aktive metabolit af Irinotecan, blev analyseret i en undergruppe af patienter (ca. 30 pr. Behandlingsarm).
SN-38-koncentrationerne var i gennemsnit 33% højere hos patienter, der fik Irinotecan / 5-FU / FA i kombination med bevacizumab sammenlignet med patienter, der fik Irinotecan / 5-FU / FA alene. På grund af den store inter-variabilitet mellem patienter og begrænset antal prøver er det usikkert, om de observerede stigninger i SN-38-niveauer var sekundære i forhold til Bevacizumab. Der var en beskeden stigning i bivirkningerne ved diarré og leukocytopeni. Større dosisreduktion af Irinotecan blev rapporteret for patienter behandlet med irinotecan / 5-FU / FA i kombination med Bevacizumab.
Hos patienter, der udvikler alvorlig diarré, leukocytopeni eller neutropeni med Bevacizumab og Irinotecan i kombination, bør dosis af Irinotecan justeres som beskrevet i pkt.4.2.
Atazanavir sulfat.
Samtidig administration af atazanavir sulfat, en hæmmer af CYP3A4 og UGT1A1, har potentiale til at øge systemisk eksponering for SN-38, den aktive metabolit af irinotecan.Læger bør tage dette i betragtning ved administration af disse lægemidler i kombination.
Interaktioner, der er fælles for alle cytotoksika: Anvendelse af antikoagulantia er almindelig på grund af en øget risiko for trombotiske hændelser i tumorsygdomme. Hvis behandling med antikoagulantia anses for at være K -vitamin -antagonister, er en større hyppighed af overvågning af INR nødvendig (International Normalized Ratio) ) på grund af deres snævre terapeutiske margen, den høje individuelle variation af blodtrombogenicitet og muligheden for interaktioner mellem orale antikoagulantia og anticancer -kemoterapi.
Samtidig brug er kontraindiceret
- Gul febervaccine: risiko for generaliseret dødelig reaktion på vacciner
Samtidig brug anbefales ikke
- Levende svækkede vacciner (undtagen gul feber): risiko for mulig dødelig systemisk sygdom (f.eks. Infektioner). Denne risiko er større hos patienter, der allerede er immunsupprimerede på grund af en allerede eksisterende sygdom.
Brug af en inaktiveret vaccine, hvor tilgængelig (poliomyelitis)
- Phenytoin: risiko for forværring af kramper som følge af faldet i intestinal absorption af phenytoin af det cytotoksiske lægemiddel. Samtidig brug skal overvejes - Ciclosporin, Tacrolimus: overdreven immunsuppression med risiko for lymfoproliferation
04.6 Graviditet og amning -
Graviditet
Der er ingen oplysninger om brugen af Irinotecan til gravide Irinotecan har vist sig at være embryotoksisk og teratogent hos dyr (se afsnit 5.3). Resultaterne af dyreforsøg og virkningsmekanismen for Iirinotecan betyder, at lægemidlet ikke gør det bør bruges under graviditet, især i første trimester, medmindre det er absolut nødvendigt. Fordelene ved behandling skal opveje de mulige risici for fosteret i hvert enkelt tilfælde.
Fertilitet
Kvinder i den fertile alder og mænd bør anvende effektiv prævention under og i mindst tre måneder efter behandlingens afslutning (se pkt. 4.4).
Der er ingen menneskelige data om virkningen af irinotecan på fertiliteten Bivirkninger af irinotecan på frugtbarhed hos afkom er registreret hos dyr (se pkt. 5.3).
Fodringstid
Det vides ikke, om Irinotecan udskilles i modermælk. 14C-Irinotecan er fundet i mælk fra diegivende rotter. På grund af potentielle bivirkninger hos spædbarnet er amning kontraindiceret under behandling med Irinotecan (se pkt. 4.3).
04.7 Virkninger på evnen til at føre motorkøretøj og betjene maskiner -
Patienter bør informeres om mulig forekomst af svimmelhed eller synsforstyrrelser, der kan forekomme inden for 24 timer efter administration af Irinotecan, og ikke at køre bil eller betjene maskiner, hvis disse symptomer opstår.
04.8 Bivirkninger -
Følgende bivirkninger er relateret til administration af Irinotecan. Der er ingen tegn på, at sikkerhedsprofilen for Irinotecan påvirkes af Cetuximab eller omvendt. I kombination med Cetuximab var de yderligere rapporterede bivirkninger de forventede bivirkninger for Cetuximab (f.eks. Acne-lignende udslæt hos 88%). Se derfor også oplysningerne om medicinen Cetuximab.
For information om bivirkninger i kombination med Bevacizumab, se Bevacizumab Resumé af produktkarakteristika.
Følgende bivirkninger, der sandsynligvis eller sandsynligvis er relateret til administration af Irinotecan, blev observeret hos 765 patienter behandlet med den anbefalede dosis på 350 mg / m² monoterapi og hos 145 patienter behandlet med Irinotecan i kombination med 5FU / FA til administration hver anden uge ved anbefalet dosis på 180 mg / m².
Bivirkninger er opsummeret i nedenstående tabel med MedDRA -frekvens. Bivirkninger præsenteres efter faldende sværhedsgrad inden for hver frekvensundergruppe.
Meget almindelig:> 1/10
Kommune:> 1/100 a
Ikke almindelig:> 1 / 1.000 y
Sjælden:> 1 / 10.000 a
Meget sjælden
De mest almindelige (> 1/10) dosisbegrænsende bivirkninger af Irinotecan er forsinket diarré (forekommer ud over 24 timer efter administration) og blodsygdomme, herunder neutropeni, anæmi og trombocytopeni.
Alvorligt forbigående akut kolinergt syndrom er generelt blevet rapporteret. Hovedsymptomer er blevet beskrevet som tidlig diarré og forskellige andre symptomer såsom mavesmerter, konjunktivitis, rhinitis, hypotension, vasodilatation, svedtendens, kuldegysninger, utilpashed, svimmelhed, synsforstyrrelser, miose, tåreflåd og øget savlen, som dukkede op inden for de første 24 timer efter infusion af Irinotecan Disse symptomer forsvandt efter administration af atropin (se pkt. 4.4).
Forsinket diarré
Ved monoterapi: Alvorlig diarré blev rapporteret hos 20% af patienterne, der fulgte anbefalingerne til kontrol af diarré. 14% af de evaluerbare cyklusser rapporterede alvorlig diarré. Den gennemsnitlige tid for begyndelsen af den første flydende afføring var den 5. dag efter infusion af Irinotecan.
I kombinationsbehandling: Alvorlig diarré blev rapporteret hos 13,1% af patienterne, der fulgte anbefalinger til behandling af diarré. Af de evaluerbare behandlingsforløb blev der rapporteret om alvorlig diarré hos 3,9%.
Blodsygdomme
Neutropeni
Neutropeni var reversibel og ikke-kumulativ; den gennemsnitlige tid til nadir var 8 dage for både monoterapi og kombinationsterapi.
Monoterapi: Neutropeni blev observeret hos 78,7% af patienterne og var alvorlig (neutrofiltal
I kombinationsbehandling: neutropeni blev observeret hos 82,5% af patienterne og var alvorlig (neutrofiltal
Feber med neutropeni blev rapporteret hos 3,4% af patienterne og 0,9% af behandlingsforløbene. Infektiøse episoder forekom hos cirka 2% af patienterne (0,5% af behandlingsforløb) og var forbundet med alvorlig neutropeni hos cirka 2,1% af patienterne (0,5% af behandlingsforløb), med dødelig udgang. I 1 tilfælde.
Anæmi
Ved monoterapi:
Anæmi blev rapporteret hos 58,7% af patienterne (8% med hæmoglobin
I kombinationsbehandling:
Anæmi blev rapporteret hos 97,2% af patienterne (2,1% med hæmoglobin
Trombocytopeni
Ved monoterapi:
Trombocytopeni (blodplader
I kombinationsbehandling:
Trombocytopeni (
Et tilfælde af perifer trombocytopeni forbundet med dannelsen af antiplatelet-antistoffer er blevet rapporteret i perioden efter markedsføring af lægemiddelovervågning.
04.9 Overdosering -
Tilfælde af overdosering er blevet rapporteret ved doser op til cirka det dobbelte af den anbefalede terapeutiske dosis, hvilket kan være dødeligt. De vigtigste rapporterede bivirkninger var alvorlig neutropeni og alvorlig diarré. Der er ingen kendt modgift mod Irinotecan. Støttende pleje bør straks startes for at forhindre dehydrering sekundært til diarré og for at behandle eventuelle infektiøse komplikationer.
05.0 FARMAKOLOGISKE EGENSKABER -
05.1 "Farmakodynamiske egenskaber -
Farmakoterapeutisk klassifikation: andre antineoplastiske midler
ATC -kode: L01XX19
Eksperimentelle data
Irinotecan er et semisyntetisk derivat af Camptothecin. Det er et antineoplastisk middel, der fungerer som en specifik hæmmer af DNA topoisomerase type I. Det metaboliseres af carboxylesterase i de fleste væv og producerer således SN-38, som er mere aktiv end Irinotecan på type I topoisomerase. renset og mere cytotoksisk end irinotecan mod forskellige mus og humane tumorcellelinjer. Inhibering af DNA topoisomerase I med irinotecan eller SN-38 inducerer enkeltstrengede DNA-læsioner, der blokerer replikationsgaffel af DNA og er ansvarlige for cytotoksicitet. Denne cytotoksiske aktivitet har blevet anerkendt som tidsafhængig og specifik for S-fasen.
In vitro, Irinotecan og SN-38 genkendes ikke af P-glycoprotein (MDR), og Irinotecan udviser cytotoksisk aktivitet mod cellelinjer, der er resistente over for Doxorubicin og Vinblastine.
Desuden har "Irinotecan" en "bred kræftaktivitet in vivo mod mus tumormodeller (pancreatisk kanal adenocarcinom P03, bryst adenocarcinom MA16 / C, colon adenocarcinomer C38 og C51) og mod humane xenotransplantater (colon adenocarcinoma Co-4, breast adenocarcinoma MX-1, gastric adenocarcinoma ST-15 og SC-16). Irinotecan er også aktivt mod tumorer, der udtrykker P-glycoprotein (MDR) (Doxorubicin og Vincristine-resistente P388 leukæmier).
Bortset fra Irinotecans anticanceraktivitet er irinotecans mest relevante farmakologiske effekt inhibering af acetylcholinesterase.
Kliniske data
Som monoterapi til andenlinjeterapi ved metastatisk kolorektal cancer
Fase II / III kliniske forsøg er blevet udført på mere end 980 patienter med metastatisk kolorektal cancer efter fejl i tidligere 5-FU-behandling med en doseringsplan hver 3. uge. Virkningen af Irinotecan blev evalueret hos 765 patienter med sygdomsprogression under dokumenteret 5FU -behandling ved studiestart.
NA: Ikke relevant
I kliniske fase II-forsøg med 455 patienter, der blev behandlet med doseringsplanen hver 3. uge, var den progressionsfrie overlevelsesrate på 6 måneder 30%, og medianoverlevelsen var 9 måneder. Mediantiden til progression var 18 uger.
Derudover blev der udført ikke-sammenlignende fase II-undersøgelser hos 304 patienter behandlet i henhold til en ugentlig tidsplan, i en dosis på 125 mg / m² administreret som en intravenøs infusion over 90 minutter i 4 på hinanden følgende uger efterfulgt af 2 ugers hvile. I disse undersøgelser var mediantiden til progressionens begyndelse 17 uger, og medianoverlevelsen var 10 måneder. En sammenlignelig sikkerhedsprofil blev observeret hos 193 patienter, der blev behandlet med den ugentlige doseringsplan ved startdosis på 125 mg / m² kontra skemaet hver tredje uge. Den gennemsnitlige tid til begyndelsen af den første væskeevakuering var 11 dage.
Kombinationsterapi til førstelinjebehandling af metastatisk kolorektal cancer
I kombinationsbehandling med folinsyre og 5-Fluorouracil
Et fase III-behandlingsstudie af første række af 385 patienter med metastatisk kolorektal cancer blev udført med lægemiddeldosering hver anden uge (se pkt. 4.2) eller en gang om ugen. Ved behandling med administration hver 2. uge, på dag 1, gives Irinotecan i en dosis på 180 mg / m² hver anden uge efterfulgt af infusion af FA (200 mg / m² som en 2-timers intravenøs infusion) og 5-FU (400 mg / m² som en intravenøs bolus efterfulgt af 600 mg / m² som en 22-timers intravenøs infusion). På dag 2 blev AF og 5-FU administreret i samme dosering og med samme skema. Ved ugentlig behandling blev dosis Irinotecan ved 80 mg / m² efterfulgt af infusion af FA (500 mg / m² som en 2-timers intravenøs infusion) og derefter 5-FU (2.300 mg / m² som en 24-timers intravenøs infusion) . i 6 uger.
I kombinationsbehandlingsstudiet med de 2 ovenfor beskrevne doseringsregimer blev effekten af Irinotecan hydrochlorid evalueret hos 198 behandlede patienter:
Irin: Irinotecan
5-FU: 5-fluorouracil,
AF: folinsyre
NS: ikke signifikant
*: i henhold til protokolpopulationsanalyse
I den ugentlige doseringsplan var "forekomsten af alvorlig diarré 44,4% hos patienter behandlet med Irinotecan i kombination med 5-FU / FA og 25,6% hos patienter behandlet med 5-FU / FA alene. L" forekomst af alvorlig neutropeni (neutrofil tælle
Desuden var den gennemsnitlige tid til at definere forringelse af ydeevnstatus signifikant længere i gruppen behandlet med Irinotecan i kombination med 5-FU / AF sammenlignet med 5-FU / AF-gruppen (p = 0,046) alene.
I dette fase III-studie blev livskvalitet undersøgt ved hjælp af EORTC QLQ-C30 spørgeskema. Tiden til begyndelsen af en endelig forringelse forekom altid senere i irinotecan -grupperne. Den generelle sundhedsstatus / livskvalitet var lidt bedre, omend ikke signifikant, i gruppen behandlet med Irinotecan i kombination, hvilket understøtter det faktum, at Irinotecans effekt i kombination kan opnås uden at gå på kompromis med livskvaliteten.
I kombinationsbehandling med cetuximab
EMR 62202-013: Denne randomiserede undersøgelse af patienter med tidligere ubehandlet metastatisk kolorektal cancer for metastatisk sygdom blev sammenlignet med kombinationen af Cetuximab og Irinotecan plus 5-fluorouracil / folinsyre (5-FU / FA) infusion (599 patienter) versus samme kemoterapi uden Cetuximab (599 patienter). Andelen af patienter med vildtype KRAS-tumorer inden for patientpopulationen, der kunne vurderes for KRAS-status, var 64%.
Effektdata genereret i denne undersøgelse er opsummeret i nedenstående tabel:
CI = konfidensinterval, FOLFIRI = Irinotecan plus 5-FU / FA infusion, ORR = objektiv responsrate (patienter med fuldstændig eller delvis respons), PFS = sygdomsfri overlevelsestid
I kombination med Cetuximab efter svigt i cytotoksisk behandling inklusive Irinotecan
Virkningen af kombinationen af Cetuximab og Irinotecan blev undersøgt i to kliniske undersøgelser: I alt 356 patienter med EFFR-udtrykkende metastatisk kolorektal cancer, hvor nylig Irinotecan-inklusiv behandling var mislykket og med Karnofsky-ydelsesstatus på mindst 60%, mens størstedelen af dem havde en Karnofsky -præstationsstatus> 80%.
EMR 62 202-007: Denne randomiserede undersøgelse sammenlignede kombinationen af Cetuximab og Irintoecan (218 patienter) med Cetuximab alene (111 patienter).
IMCL CP02-9923: Denne åbne enkeltarmstudie undersøgte kombinationsbehandling hos 138 patienter.
Effektdataene fra disse undersøgelser er opsummeret nedenfor.
CI = konfidensinterval; DCR = sygdomskontrolrate (patienter med fuldstændig eller delvis respons eller stationær sygdom i mindst 6 uger); ORR = objektiv responsrate (patienter med fuldstændig eller delvis respons); OS = samlet overlevelsestid; PFS progressionsfri overlevelse
Med hensyn til hyppigheden af objektiv respons (ORR), sygdomsbekæmpelse (DCR) og sygdomsfri overlevelse (PFS) var effekten af kombinationen af Cetuximab og Irinotecan højere end Cetuximabs alene.I det randomiserede studie er de ikke effekter på den samlede overlevelse er blevet påvist (hazard ratio 0,91, p = 0,48).
I kombinationsterapi med Bevacizumab
Et dobbeltblindet, randomiseret, kontrolleret fase III klinisk forsøg evaluerede bevacizumab i kombination med Irinotecan / 5-FU / FA som førstelinjebehandling for metastatisk kolorektal cancer (undersøgelse AVD2170g). Tilsætningen af Bevacizumab til kombinationen Irinotecan / 5-FU / FA resulterede i en statistisk signifikant stigning i den samlede overlevelse. Klinisk fordel målt ved samlet overlevelse var tydelig i alle præspecificerede patientundergrupper, herunder dem defineret efter alder., køn, præstationsstatus, primær tumorplacering, antal involverede organer og varighed af metastatisk sygdom. Se også Bevacizumab Resumé af produktkarakteristika. Tabellen nedenfor opsummerer effektivitetsresultaterne fra undersøgelse AVF2107g.
Farmakokinetiske / farmakodynamiske data
Intensiteten af de vigtigste toksiske virkninger set med irinotecanhydrochlorid (f.eks. Diarré og neutropeni) er relateret til eksponeringen (AUC-området under kurven) for modermedicinen og metabolitten SN-38. Der blev observeret en signifikant sammenhæng mellem hæmatologisk toksicitet (nedsat leukocytter og neutrofiler ved nadir) eller mellem intensiteten af diarré og AUC-værdier for både irinotecan og metabolitten SN-38 alene.
Patienter med nedsat UGT1A1-aktivitet: Uridindiphosphat-glucuronosyl 1A1-transferase (UGT1A1) er involveret i metabolisk deaktivering af SN-38, den aktive metabolit af irinotecan, til det inaktive SN-38-glucuronid (SN-38G). UGT1A1-genet er meget polymorf, hvilket resulterer i variabel metabolisk kapacitet mellem individer. En specifik variant af UGT1A1-genet inkluderer en polymorfisme i promotorregionen kendt som UGT1A1 * 28-varianten. Denne variant og andre medfødte mangler ved UGT1A1-udtryk (såsom Crigler-Najjar og Gilbert) er forbundet med en reduceret aktivitet af dette enzym.
Metaanalysedata viser, at personer med Crigler-Najjar syndrom (type 1 og 2) eller homozygote for UGT1A1 * 28-allelet (Gilbert syndrom) har øget risiko for hæmatologisk toksicitet (grad 3 og 4). Efter administration af irinotecan ved moderat eller høje doser (> 150 mg / m²). Et forhold mellem UGT1A1-genotypen og forekomsten af irinotecan-induceret diarré er ikke blevet fastslået. Patienter, der vides at være homozygote for UGT1A1 * 28, bør behandles med den normalt indikerede startdosis irinotecan Imidlertid bør disse patienter overvåges for hæmatologisk toksicitet.En reduceret startdosis af irinotecan bør overvejes for patienter, der har oplevet hæmatologisk toksicitet ved tidligere behandling. bør være baseret på tolerance over for patientbehandling. (se afsnit 4.2 og 4.4) Der er i øjeblikket utilstrækkelige data til at drage konklusioner om den kliniske anvendelighed af UGT1A1 genotyping.
05.2 "Farmakokinetiske egenskaber -
I et fase I-studie med 60 patienter med en 30 minutters intravenøs infusionsdosis på 100 til 750 mg / m² en gang hver tredje uge udviste Irinotecan en bifasisk eller trifasisk eliminationsprofil.Den gennemsnitlige plasmaclearance var 15 l / h / m², og distributionsvolumen ved steday-tilstand (Vdss) var 157 l / m². Plasmahalveringstiden for den første fase af den trifasiske model var 12 minutter, den for den anden fase var 2,5 timer, og terminalfasehalveringstiden var 14,2 timer. SN-38 udviste en bifasisk eliminationsprofil med en gennemsnitlig terminal eliminationshalveringstid på 13,8 timer. Ved afslutningen af infusionen, ved den anbefalede dosis på 350 mg / m², var de gennemsnitlige maksimale plasmakoncentrationer af Irinotecan og SN-38 henholdsvis 7,7 mcg / ml og 56 ng / ml med tilsvarende middelværdier for "areal" under kurven (AUC) på henholdsvis 34 mcg.h / ml og 451 ng.h / ml. En "bred interindividuel variation af farmakokinetiske parametre blev observeret især for SN-38".
En "farmakokinetisk analyse af irinotecan" blev udført i en population på 148 patienter med metastatisk kolorektal cancer behandlet med forskellige skemaer og med forskellige doser i fase II -undersøgelser. De farmakokinetiske parametre beregnet i tre-rums-modellen lignede meget dem, der blev observeret i fase I-undersøgelserne. Alle undersøgelser viste, at eksponering for Irinotecan (CPT-11) og SN-38 stiger proportionalt med administreret dosis CPT-11; deres farmakokinetik er uafhængige af antallet af tidligere cyklusser og af behandlingsregimet.
Bindingen til plasmaproteiner in vitro, af Irinotecan og SN-38 var henholdsvis ca. 65% og 95%.
Massebalance og metaboliske undersøgelser udført med det 14-C-mærkede lægemiddel har vist, at mere end 50% af en intravenøst administreret dosis Irinotecan udskilles uændret, 33% udskilles i fæces hovedsageligt med galde. Og 22% i urinen.
To metaboliske veje er hver ansvarlig for mindst 12% af dosis:
• Carboxylesterase-medieret hydrolyse for at aktivere den aktive metabolit SN-38. SN-38 elimineres primært ved glucuronidering og udskilles yderligere via galde- og nyrevejene (mindre end 0,5% af Irinotecan-dosis). SN-38-glucuronid hydrolyseres sandsynligvis efterfølgende i tarmen.
• Oxidation fremmet af enzymet P450 3A, hvilket resulterer i åbning af den ydre ring af piperidin med dannelse af derivatet af aminopentansyre (PCA) og primært aminderivat (NPC) (se afsnit 4.5).
I plasma er den største enhed uændret Irinotecan, efterfulgt af APC, SN-38-glucuronid og SN-38. Kun SN-38 har en betydelig cytotoksisk virkning.
Irinotecans clearance reduceres med ca. 40% hos patienter med bilirubin mellem 1,5 og 3 gange den øvre grænse for det normale område. Hos disse patienter resulterer en dosis Irinotecan på 200 mg / m² i en plasmaeksponering af lægemidlet, der kan sammenlignes med det, der blev fundet for 350 mg / m² hos kræftpatienter med normale leverparametre.
05.3 Prækliniske sikkerhedsdata -
Irinotecan og SN-38 har vist sig at være mutagene in vitro i den kromosomale aberrationstest på CHO -celler samt in vivo i musens mikronukleustest. I Ames -testen viste de sig imidlertid at være blottet for ethvert mutagent potentiale.
Hos rotter, der blev behandlet en gang om ugen i 13 uger med den maksimale dosis på 150 mg / m² (hvilket er mindre end halvdelen af den anbefalede humane dosis), blev der ikke rapporteret om behandlingsrelaterede tumorer inden for den 91-ugers periode derefter. Terapi.
Enkelt- og gentagen dosis toksicitetsundersøgelser blev udført på mus, rotter og hunde. De vigtigste toksiske virkninger blev observeret i de hæmatopoietiske og lymfatiske systemer. Forsinket diarré forbundet med fokal atrofi og nekrose af tarmslimhinden er blevet rapporteret hos hunde. Alopeci blev også observeret hos hunde. Alvorligheden af disse virkninger er dosisrelateret og reversibel.
06.0 LÆGEMIDDELOPLYSNINGER -
06.1 Hjælpestoffer -
Sorbitol (E420)
Mælkesyre (E270)
Natriumhydroxid og / eller saltsyre (for at justere pH)
Vand til injektionsvæsker
06.2 Uforenelighed "-
Dette lægemiddel må ikke blandes med andre medicinske produkter undtagen dem, der er nævnt i afsnit 6.6.
06.3 Gyldighedsperiode "-
Holdbarheden for uåbnede hætteglas er 3 år.
Hætteglas med Irinotecan til infusion skal bruges straks efter åbning, da de ikke indeholder antimikrobielle konserveringsmidler.
Stabilitet efter fortynding:
Kemisk-fysisk stabilitet ved brug er blevet påvist i glucose 50 mg / ml (5%) og i natriumchlorid 9 mg / ml (0,9%) i 72 timer mellem 2 ° C og 8 ° C. Fra et mikrobiologisk synspunkt bør produktet bruges med det samme. Hvis den ikke bruges med det samme, er opbevaringstider og -betingelser inden brug brugerens ansvar og vil normalt ikke overstige 24 timer ved 2 ° C til 8 ° C, medmindre fortynding foretages under aseptiske forhold. Kontrolleres og valideres.
06.4 Særlige opbevaringsforhold -
Opbevar hætteglasset i den originale emballage. Må ikke fryses.
Hætteglas med Irinotecan hydrochlorid koncentrat til infusionsvæske, opløsning skal beskyttes mod lys.
Opbevaringsbetingelser for det fortyndede lægemiddel henvises til pkt. 6.3.
06.5 Den umiddelbare emballages art og emballagens indhold -
• 40 mg / 2 ml: Et 5 ml Onco-Tain hætteglas af type I med fluorobutylgummilukning dækket med teflon på indersiden.
• 100 mg / 5 ml: Et 5 ml brunt Onco-tain hætteglas af type I med et luorobutylgummilukning dækket med teflon på indersiden.
• 500 mg / 25 ml: Et Onco-Tain Type I brunt 30 ml hætteglas med fluorobutylgummilukning dækket med teflon på indersiden.
Hver pakning indeholder et hætteglas. Ikke alle godkendte og fremhævede pakningsstørrelser markedsføres muligvis.
Onco-Tain er Hospiras proprietære eksterne hætteglasbeskyttelsessystem.
06.6 Brugsanvisning og håndtering -
Opløsningen skal fortyndes før brug. Emballager til engangsbrug. Eventuelle rester i hætteglasset skal kasseres.
Som med andre antineoplastiske lægemidler bør Irinotecan -infusioner tilberedes og håndteres med omhu. Brug af beskyttelsesbriller, maske og handsker er påkrævet. Gravide bør ikke håndtere cytotoksiske stoffer.
Hvis opløsningen af Irinotecan -koncentrat til infusion eller infusionsvæsken skal komme i kontakt med huden, vaskes straks med rigeligt vand og sæbe. I kontakt med slimhinder, vaskes straks med vand.
Forberedelse til administration af intravenøs infusion: Som alle andre injicerbare lægemidler skal Irinotecan -opløsningen tilberedes aseptisk (se afsnit 6.3).
Hvis der observeres et bundfald i hætteglassene eller infusionsvæsken, skal produktet kasseres efter standard hospitalsprocedurer, der gælder for cytotoksiske lægemidler.
Under aseptiske forhold trækkes den nødvendige mængde af den koncentrerede Irinotecan -opløsning op fra hætteglasset ved hjælp af en gradueret sprøjte og injiceres i en 250 ml infusionspose eller flaske, der kun indeholder natriumchlorid 9 mg / ml (0,9%) eller glucoseopløsning 50 mg / ml ( 5%). Infusionen skal derefter blandes perfekt ved manuel rotation.
Eliminering. Alt materiale, der bruges til fortynding og administration, skal kasseres i henhold til standard hospitalsprocedurer, der gælder for cytotoksiske lægemidler.
07.0 INDEHAVER AF "MARKEDSFØRINGSTILLADELSEN" -
Hospira Italia S.r.l.
Via Orazio, 20/22
80122 Napoli
Italien
08.0 MARKEDSFØRINGSTILLADELSESNUMMER -
Irinotecan Hospira 20 mg / ml koncentrat til infusionsvæske, opløsning 2 ml hætteglas
A.I.C. n. 037037013
Irinotecan Hospira 20 mg / ml koncentrat til infusionsvæske, opløsning 5 ml hætteglas
A.I.C. n. 037037025
Irinotecan Hospira 20 mg / ml koncentrat til infusionsvæske, opløsning i 25 ml hætteglas
A.I.C. n. 037037037
09.0 DATO FOR FØRSTE TILLADELSE ELLER FORNYELSE AF TILLADELSEN -
Bestemmelse n. 258 af 19. juli 2006
Lovtidende nr. 178 af 2. august 2006
10.0 DATO FOR REVISION AF TEKSTEN -
02/2014