Aktive ingredienser: Bevacizumab
Avastin 25 mg / ml koncentrat til infusionsvæske, opløsning
Hvorfor bruges Avastin? Hvad er det for?
Avastin indeholder det aktive stof bevacizumab, et humaniseret monoklonalt antistof (generelt er antistoffer en type protein, der normalt produceres af immunsystemet for at hjælpe kroppen til at forsvare sig mod infektion og kræft).
Bevacizumab binder selektivt til et protein kaldet 'human vaskulær endotelvækstfaktor' (VEGF), som er til stede på foringen af kroppens blod og lymfekar. VEGF -proteinet bestemmer væksten af blodkar i tumoren; disse blodkar forsyner tumoren med næringsstoffer og ilt. Når bevacizumab binder sig til VEGF, forhindres tumorvækst ved at blokere udviklingen af blodkar, der leverer næringsstoffer. Og ilt til tumoren Avastin er en medicin, der bruges til behandling af voksne patienter med fremskreden kræft i tyktarmen, dvs. tyktarm eller endetarm. Avastin vil blive givet i kombination med en kemoterapibehandling indeholdende en fluoropyrimidinbaseret medicin.
Avastin bruges også til behandling af voksne patienter med metastatisk brystkræft. Hos patienter med denne type kræft vil Avastin blive givet med et paclitaxel- eller capecitabinbaseret kemoterapiregime.
Avastin bruges også til behandling af voksne patienter med fremskreden ikke-småcellet lungekræft. Avastin vil blive givet sammen med et platinbaseret kemoterapiregime.
Avastin bruges også til behandling af voksne patienter med fremskreden nyrekræft. Hos patienter med denne type kræft vil Avastin få en anden type medicin kaldet interferon.
Avastin bruges også til behandling af voksne patienter med epitelial æggestokkræft, æggelederkræft eller fremskreden primær peritoneal kræft. Hos patienter med denne type kræft vil Avastin blive givet i kombination med carboplatin og paclitaxel.
Avastin vil blive givet i kombination med carboplatin og gemcitabin, når det bruges til voksne patienter med epitelial æggestokkræft, æggelederkræft eller fremskreden primær peritoneal kræft, hvis sygdom har manifesteret sig igen mindst 6 måneder efter sidste gang, de blev behandlet med et kemoterapiregime, der indeholder et platinbaseret middel.
Avastin vil blive givet i kombination med paclitaxel, topotecan eller pegyleret liposomal doxorubicin, når det bruges til voksne patienter med epitelial æggestokkræft, æggelederkræft eller fremskreden primær peritoneal kræft, hvis sygdom er genopstået mindre end 6 måneder efter den sidste. behandlet med en kemoterapibehandling indeholdende et platinbaseret middel.
Avastin bruges også til behandling af voksne patienter med vedvarende, tilbagevendende eller metastatisk livmoderhalskræft.Avastin vil blive givet i kombination med paclitaxel og cisplatin eller alternativt paclitaxel og topotecan til patienter, der ikke kan behandles med platin.
Kontraindikationer Når Avastin ikke bør bruges
Brug ikke Avastin:
- hvis du er allergisk (overfølsom) over for bevacizumab eller et af de øvrige indholdsstoffer i denne medicin
- hvis du er allergisk (overfølsom) over for produkter afledt af kinesiske hamsterceller (CHO) eller andre humane eller humaniserede rekombinante antistoffer.
- hvis du er gravid.
Forholdsregler ved brug Det, du skal vide, før du tager Avastin
Tal med din læge, apotek eller sygeplejerske, før du bruger Avastin
- Det er muligt, at Avastin kan øge risikoen for at udvikle perforeringer i tarmvæggen. Hvis du har tilstande, der forårsager betændelse i maven (f.eks. Diverticulitis, mavesår, kemoterapi -associeret colitis), skal du diskutere dette med din læge.
- Avastin kan øge risikoen for at udvikle en unormal forbindelse eller passage mellem to organer eller kar. Tilstedeværelsen af vedvarende, tilbagevendende eller metastatisk livmoderhalskræft kan føre til en øget risiko for at udvikle forbindelser mellem skeden og enhver del af mave -tarmkanalen.
- Denne medicin kan øge risikoen for blødning eller øge risikoen for problemer med sårheling efter operationen. Hvis du skal opereres, hvis du har været igennem en større operation i de sidste 28 dage, eller hvis du har et kirurgisk sår, der endnu ikke er helet, bør du ikke tage denne medicin.
- Avastin kan øge din risiko for at udvikle alvorlige infektioner i huden eller dybere lag under huden, især hvis du har perforeringer i tarmvæggen eller har problemer med sårheling.
- Avastin kan øge forekomsten af forhøjet blodtryk. Hvis du har forhøjet blodtryk, der ikke er godt kontrolleret med blodtryksmedicin, skal du diskutere dette med din læge. Det er vigtigt at sikre dig, at dit blodtryk er under kontrol, inden du starter behandling med Avastin.
- Denne medicin øger risikoen for at have protein i din urin, især hvis du allerede har forhøjet blodtryk.
- Risikoen for at udvikle blodpropper i arterierne (en type blodkar) kan stige, hvis du er over 65 år, har diabetes og tidligere har haft blodpropper i arterierne. Tal med din læge, fordi blodpropper kan føre til hjerteanfald og slagtilfælde.
- Avastin kan også øge risikoen for at udvikle blodpropper i venerne (en type blodkar).
- Denne medicin kan forårsage blødning, især tumorrelateret blødning. Kontakt din læge, hvis du eller andre familiemedlemmer har en tendens til at have problemer med blodpropper, eller hvis du tager blodfortyndende medicin af en eller anden grund.
- Det er muligt, at Avastin kan forårsage blødning i og omkring hjernen. Kontakt din læge, hvis du har metastatisk sygdom, der involverer hjernen.
- Det er muligt, at Avastin kan øge risikoen for blødning i lungerne, herunder blod i hosten eller spyt. Tal med din læge, hvis du tidligere har bemærket disse hændelser.
- Avastin kan øge risikoen for at udvikle "hjertesvigt. Det er vigtigt for din læge at vide, om du tidligere har modtaget antracykliner (f.eks. Doxorubicin, en særlig type kemoterapi, der bruges til behandling af nogle kræftformer) eller strålebehandling af brystet. Eller hvis du har hjerte sygdom.
- Denne medicin kan forårsage infektioner og reducere antallet af neutrofiler (en type blodlegemer, der er vigtig for beskyttelse mod bakterier).
- Det er muligt, at Avastin kan forårsage overfølsomhed og / eller infusionsreaktioner (reaktioner relateret til injektion af medicinen) Fortæl det til din læge, apotek eller sygeplejerske, hvis du allerede har haft problemer efter injektionerne, såsom svimmelhed / besvimelse, mangel på vejrtrækning, hævelse eller udslæt.
- En sjælden neurologisk bivirkning kaldet posterior reversibel encephalopathysyndrom er blevet forbundet med behandling med Avastin. Hvis du har hovedpine, forstyrret syn, forvirring eller anfald med eller uden forhøjet blodtryk, skal du kontakte din læge.
Tal med din læge, selvom ovenstående kun er sket tidligere.
Inden behandling påbegyndes med Avastin eller under behandling med Avastin:
- hvis du har haft eller har smerter i munden, tænderne og / eller kæben eller hævelse eller betændelse i munden eller følelsesløshed eller tyngde i kæben eller taber en tand, skal du straks rapportere det til din læge og tandlæge;
- fortæl din tandlæge, at du er i behandling med Avastin, især hvis du har modtaget eller modtager en "bisphosphonat -injektion. Din læge eller tandlæge kan foreslå, at du får en tandtjek -op, før du går i gang med behandling med Avastin.
Interaktioner Hvilke lægemidler eller fødevarer kan ændre virkningen af Avastin
Fortæl det til din læge, apotek eller sygeplejerske, hvis du tager anden medicin eller har brugt det for nylig.
Kombination af Avastin med en anden medicin kaldet sunitinibmalat (ordineret til nyre- og mave -tarmkræft) kan forårsage alvorlige bivirkninger. Tal med din læge for at sikre, at du ikke kombinerer disse lægemidler.
Fortæl det til din læge, hvis du bruger platin- eller taxanbaserede terapier til metastatisk lunge- eller brystkræft. Disse behandlinger i kombination med Avastin kan øge risikoen for alvorlige bivirkninger.
Fortæl det til din læge, hvis du for nylig har modtaget eller i øjeblikket modtager strålebehandling.
Advarsler Det er vigtigt at vide, at:
Børn og unge
Avastinbehandling anbefales ikke til børn og unge under 18 år, da hverken sikkerhed eller fordele er fastslået i denne patientpopulation.
Giv ikke Avastin til børn i alderen 3 til 18 med ondartede tumorer i hjernen og rygmarven, der vokser hurtigt og udvikler sig gennem hjernevæv efter behandlingssvigt (tilbagefald eller progressivt gliom af høj kvalitet), da to begrænsede undersøgelser har vist ineffektivitet i disse typer af tumorer.
Graviditet, amning og fertilitet
Hvis du er gravid, bør du ikke bruge Avastin. Avastin kan skade det ufødte barn, da det kan stoppe dannelsen af nye blodkar. Din læge vil råde dig til at bruge passende prævention under Avastin -behandlingen og i mindst 6 måneder efter at have taget den sidste dosis Avastin.
Hvis du er gravid, hvis du har mistanke om, at du er gravid, mens du tager denne medicin eller planlægger at blive gravid i den nærmeste fremtid, skal du straks tale med din læge.
Du bør ikke amme din baby, mens du tager Avastin og i mindst 6 måneder efter at have taget den sidste dosis Avastin, da Avastin kan forstyrre din babys vækst og udvikling.
Avastin kan reducere kvindelig fertilitet. Kontakt din læge for mere information.
Spørg din læge, apotek eller sygeplejerske til råds, før du tager medicin.
Kørsel og brug af maskiner
Avastin har ikke vist sig at reducere evnen til at føre motorkøretøj eller bruge værktøjer eller maskiner. Dog er der rapporteret søvnighed og synkope ved brug af Avastin. Hvis du oplever symptomer, der påvirker dit syn eller din koncentration, eller din reaktionsevne, må du ikke køre bil eller betjene maskiner, før symptomerne forsvinder.
Dosis, metode og administrationstidspunkt Sådan bruges Avastin: Dosering
Dosering og hyppighed af administration
Den dosis Avastin, du har brug for, afhænger af din kropsvægt og den kræftform, der behandles. Den anbefalede dosis er 5 mg, 7,5 mg, 10 mg eller 15 mg pr. Kg legemsvægt. Din læge vil ordinere Avastin i den passende dosis til dig. Behandling med Avastin vil blive givet dig en gang hver 2. til 3. uge. Antallet af infusioner, du modtager, afhænger af din reaktion på behandlingen; du skal dog fortsætte behandlingen, indtil Avastin ikke længere kan stoppe din tumor fra at vokse. Din læge vil ikke tale med dig.
Metode og indgivelsesvej
Avastin er et koncentrat til infusionsvæske, opløsning. Afhængigt af den dosis, der er ordineret til dig, vil en del af indholdet i Avastin -hætteglasset eller hele hætteglasset blive fortyndet med natriumchloridopløsning før brug. Din læge eller sygeplejerske vil give dig denne fortyndede opløsning af Avastin som en intravenøs infusion (et dryp i en vene). Den første infusion vil blive givet over 90 minutter. Hvis dette tolereres godt, kan den anden infusion gives i løbet af 60 minutter. Efterfølgende infusioner kan gives til dig i løbet af 30 minutter.
Administration af Avastin bør afbrydes midlertidigt
- hvis du udvikler alvorlige problemer med forhøjet blodtryk, som kræver behandling med medicin til at kontrollere dit blodtryk,
- hvis du har sårhelingsproblemer efter operationen,
- hvis du skal til en "operation".
Administration af Avastin skal afbrydes permanent, hvis et af følgende problemer opstår
- alvorligt forhøjet blodtryk, der ikke kan kontrolleres med passende medicin, eller pludselig og alvorlig stigning i blodtrykket,
- tilstedeværelse af protein i urinen forbundet med ødem (hævelse af kroppen),
- perforering af tarmvæggen,
- en unormal forbindelse eller passage mellem luftrøret og spiserøret, indre organer og hud, skeden og enhver del af mave -tarmkanalen eller mellem andre væv, der normalt ikke er forbundet (fistel), og som af lægen vurderes at være svære,
- alvorlige infektioner i huden eller de dybere lag under huden,
- blodpropper i arterierne,
- blodpropper i lungens blodkar,
- alvorlig blødning af enhver art.
Overdosering Hvad skal man gøre, hvis man har taget for meget Avastin
Hvis der gives for meget Avastin
- Du kan opleve alvorlig hovedpine. I dette tilfælde skal du straks kontakte din læge, apotek eller sygeplejerske.
Hvis du har glemt at tage en dosis Avastin
- Din læge vil beslutte, hvornår det er bedst for dig at tage din næste dosis Avastin. Diskuter dette med din læge.
Hvis du holder op med at tage Avastin
Afbrydelse af behandlingen med Avastin kan stoppe tumorvækstens begrænsning. Stop ikke med at tage Avastin, før du har talt med din læge.
Spørg din læge, apotek eller sygeplejerske, hvis du har yderligere spørgsmål om brugen af dette lægemiddel.
Bivirkninger Hvad er bivirkningerne af Avastin
Ligesom al anden medicin kan denne medicin forårsage bivirkninger, men ikke alle får bivirkninger.
Tal med din læge, apotek eller sygeplejerske, hvis du får bivirkninger. Dette omfatter eventuelle bivirkninger, der ikke er anført i denne indlægsseddel.
Nedenstående bivirkninger er blevet observeret hos patienter behandlet med Avastin kombineret med kemoterapi. Dette betyder ikke, at disse bivirkninger nødvendigvis var forårsaget af Avastin.
Allergiske reaktioner
Hvis du har en allergisk reaktion, skal du straks fortælle det til din læge eller et medlem af det medicinske personale. Tegnene kan omfatte: vejrtrækningsbesvær eller brystsmerter. Der kan også være rødme i huden eller rødme eller udslæt, kuldegysninger og rysten, kvalme eller opkastning.
Hvis du oplever nogen af de bivirkninger, der er beskrevet nedenfor, skal du straks få hjælp.
Alvorlige bivirkninger, som kan være meget almindelige (rammer mere end 1 ud af 10 patienter), omfatter:
- højt blodtryk,
- følelse af følelsesløshed eller prikken i hænder eller fødder,
- reduktion i antallet af blodlegemer, herunder hvide blodlegemer, der virker mod infektioner (dette kan ledsages af feber) og i de celler, der bidrager til blodpropper,
- følelse af svaghed og mangel på energi,
- træthed,
- diarré, kvalme, opkastning og mavesmerter.
Alvorlige bivirkninger, som kan være almindelige (rammer 1 til 10 brugere ud af 100), omfatter:
- tarmperforering,
- blødning, herunder blødning i lungerne hos patienter med ikke-småcellet lungekræft,
- arterier blokeret af en blodprop,
- vener blokeret af en blodprop,
- lunge blodkar blokeret af en blodprop,
- benårer blokeret af en blodprop,
- hjertefejl,
- sårhelingsproblemer efter operationen,
- rødme, afskalning, ømhed, smerter eller blærer i fingre eller fødder,
- reduktion i antallet af røde blodlegemer,
- mangel på energi,
- mave- og tarmsygdomme,
- muskel- og ledsmerter, muskelsvaghed,
- mundtørhed forbundet med tørst og / eller nedsat eller mørk urin,
- betændelse i mundslimhinden, tarmene, lungerne og luftvejene, reproduktive og urinveje,
- sår i munden og spiserøret, som kan forårsage smerter og synkebesvær,
- smerter, herunder hovedpine, rygsmerter og smerter omkring bækkenet og anus,
- lokaliserede bylder,
- infektion, og især infektion i blodet eller blæren,
- nedsat blodtilførsel til hjernen eller slagtilfælde
- døsighed,
- næseblod,
- øget puls (puls),
- tarmblokering,
- unormale urinprøver (tilstedeværelse af protein i urinen),
- åndenød eller nedsat iltindhold i blodet
- infektioner i huden eller dybere lag af huden,
- fistler: unormal rørformet forbindelse mellem indre organer og hud eller andre væv, der normalt ikke er forbundet med hinanden, herunder forbindelser mellem vagina og mave -tarmkanalen hos patienter med livmoderhalskræft.
Alvorlige bivirkninger af ukendt frekvens (frekvensen kan ikke estimeres ud fra de tilgængelige data) omfatter:
- alvorlige hudinfektioner eller dybere lag under huden, især hvis du har haft perforeringer i tarmvæggen eller problemer med sårheling,
- allergiske reaktioner (tegn kan omfatte vejrtrækningsbesvær, rødme i ansigtet, udslæt, lavt eller forhøjet blodtryk, lav ilt i blodet, smerter i brystet eller kvalme / opkastning),
- en negativ indvirkning på kvinders evne til at få børn (se de næste afsnit i listen over bivirkninger for yderligere anbefalinger),
- en tilstand i hjernen med symptomer som anfald (anfald), hovedpine, forvirring og ændringer i synet (posterior reversibel encefalopati syndrom (PRES)),
- symptomer, der tyder på ændringer i normal hjernefunktion (hovedpine, synsforstyrrelser, forvirring eller anfald) og forhøjet blodtryk,
- obstruktion af et eller flere små blodkar i nyrerne,
- et "unormalt højt blodtryk i lungerne, hvilket får den højre side af hjertet til at arbejde hårdere end normalt,
- perforering af bruskvæggen, der adskiller næseborene,
- perforering af maven eller tarmene,
- et åbent sår eller perforering i slimhinden i maven eller tyndtarmen (tegn kan omfatte mavesmerter, oppustethed, sort tarry afføring, blod i afføring eller blod i opkast),
- blødning fra den nederste del af tyktarmen,
- tandkødsskade med eksponering af et ikke-helbredende kæbeknog, som kan være forbundet med smerter og betændelse i det omgivende væv (se de følgende afsnit i listen over uønskede virkninger for yderligere anbefalinger),
- galdeblæreperforering (symptomer og tegn kan omfatte mavesmerter, feber og kvalme / opkastning).
Hvis du oplever nogen af de bivirkninger, der er beskrevet nedenfor, skal du få hjælp hurtigst muligt
Meget almindelige bivirkninger (forekommer hos mere end 1 ud af 10 patienter), som ikke var alvorlige, omfatter:
- forstoppelse,
- mistet appetiten,
- feber,
- øjenproblemer (herunder øget rivning),
- taleændringer,
- ændret smagssans,
- en løbende næse,
- tør hud, skrælning og betændelse i huden, ændring i hudfarve,
- tab af kropsvægt.
Almindelige bivirkninger (påvirker 1 til 10 brugere ud af 100), som ikke var alvorlige, omfatter:
- stemmeændringer og hæshed.
Patienter over 65 år har en øget risiko for at få følgende bivirkninger:
- blodpropper i arterierne, hvilket kan føre til slagtilfælde eller hjerteanfald
- reduktion i antallet af hvide blodlegemer og celler, der bidrager til blodpropper,
- diarré,
- følelse af utilpashed,
- hovedpine,
- følelse af træthed,
- højt blodtryk.
Avastin kan også forårsage ændringer i resultaterne af de laboratorietests, som din læge har ordineret. Disse omfatter: en reduktion i antallet af hvide blodlegemer, især neutrofiler (en type hvide blodlegemer, der hjælper med at beskytte mod infektion) i blodet, tilstedeværelsen af protein i urinen, et fald i kalium, natrium eller fosfor (et mineral) i blodet, forhøjet blodsukker, forhøjet alkalisk fosfatase (et enzym) i blodet, nedsat hæmoglobin (findes i røde blodlegemer og transporterer ilt), hvilket kan være alvorligt.
Smerter i munden, tænder og / eller kæbe, hævelse eller blærer i munden, følelsesløshed eller en følelse af tyngde i kæben eller løsn af en tand. Disse kan være tegn og symptomer på knogleskade i kæben (osteonekrose).Fortæl det straks til din læge og tandlæge, hvis nogen af disse virkninger opstår.
Kvinder før overgangsalderen (kvinder, der har en menstruationscyklus) kan mærke uregelmæssige menstruationscyklusser, mangel på menstruation og kunne have negative konsekvenser for fertiliteten. Hvis du overvejer at få børn, bør du diskutere dette med din læge, inden du starter behandlingen.
Avastin blev udviklet og fremstillet til behandling af kræft ved intravenøs injektion.
Det blev ikke udviklet eller fremstillet til administration ved injektion i øjet.
Derfor er brugen af denne indgivelsesvej ikke tilladt. Når Avastin injiceres direkte i øjet (brug ikke godkendt), kan følgende bivirkninger forekomme:
- infektion eller betændelse i øjeæblet,
- øjenrødme, partikler eller flydende punkter i synsfeltet ("flyvende fluer"), smerter i øjet,
- lysglimt og "flyvende fluer", der udvikler sig til tab af en del af synsfeltet,
- øget tryk i øjet,
- øjenblødning.
Indberetning af bivirkninger
Tal med din læge, apotek eller sygeplejerske, hvis du får bivirkninger. Dette omfatter eventuelle bivirkninger, der ikke er anført i denne indlægsseddel. Du kan også rapportere bivirkninger direkte via det nationale rapporteringssystem anført i tillæg V.
Ved at rapportere bivirkninger kan du hjælpe med at give flere oplysninger om sikkerheden ved dette lægemiddel.
Udløb og opbevaring
Opbevar denne medicin utilgængeligt for børn.
Brug ikke dette lægemiddel efter den udløbsdato, der står på den ydre karton og hætteglassets etiket efter forkortelsen EXP. Udløbsdatoen refererer til den sidste dag i måneden.
Opbevares i køleskab (2 ° C-8 ° C).
Må ikke fryses
Opbevar hætteglasset i den ydre karton for at beskytte medicinen mod lys.
Infusionsopløsninger skal bruges umiddelbart efter fortynding. Brug ikke Avastin, hvis du bemærker partikler eller farveændringer inden administration.
Smid ikke medicin via spildevand eller husholdningsaffald. Spørg din apotek om, hvordan du skal smide medicin, du ikke længere bruger. Dette vil hjælpe med at beskytte miljøet.
Pakningens indhold og andre oplysninger
Hvad Avastin indeholder
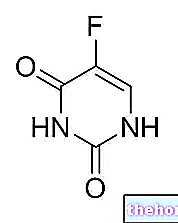
- Den aktive ingrediens er bevacizumab.
Hver ml koncentrat indeholder 25 mg bevacizumab, svarende til 1,4-16,5 mg / ml, når det fortyndes som anbefalet.
Hvert 4 ml hætteglas indeholder 100 mg bevacizumab, svarende til 1,4 mg / ml, når det fortyndes som anbefalet.
Hvert 16 ml hætteglas indeholder 400 mg bevacizumab, svarende til 16,5 mg / ml, når det fortyndes som anbefalet.
- Øvrige indholdsstoffer er trehalosedihydrat, natriumphosphat, polysorbat 20 og vand til injektionsvæsker.
Hvordan Avastin ser ud og pakningens indhold
Avastin er et koncentrat til infusionsvæske, opløsning. Koncentratet er en klar, farveløs til lysebrun væske i et hætteglas med glas lukket med en gummiprop. Hvert hætteglas indeholder 100 mg bevacizumab i 4 ml opløsning eller 400 mg bevacizumab i 16 ml opløsning. Hver Avastin -pakning indeholder et hætteglas.
Indlægsseddel: AIFA (Italian Medicines Agency). Indhold offentliggjort i januar 2016. De foreliggende oplysninger er muligvis ikke opdaterede.
For at få adgang til den mest opdaterede version er det tilrådeligt at få adgang til webstedet AIFA (Italian Medicines Agency). Ansvarsfraskrivelse og nyttige oplysninger.
01.0 LÆGEMIDLETS NAVN
AVASTIN 25 MG / ML KONCENTRAT TIL INFUSIONSLØSNING
02.0 KVALITATIV OG KVANTITATIV SAMMENSÆTNING
Hver ml koncentrat indeholder 25 mg bevacizumab *.
Hvert 4 ml hætteglas indeholder 100 mg bevacizumab.
Hvert 16 ml hætteglas indeholder 400 mg bevacizumab.
For fortynding og andre håndteringsanbefalinger, se afsnit 6.6.
* Bevacizumab er et humaniseret monoklonalt antistof fremstillet ved rekombinant DNA -teknik i ovarieceller fra kinesisk hamster.
Den fulde liste over hjælpestoffer findes i afsnit 6.1.
03.0 LÆGEMIDDELFORM
Koncentrat til infusionsvæske, opløsning.
Klar til let opaliserende og farveløs til lysebrun væske.
04.0 KLINISKE OPLYSNINGER
04.1 Terapeutiske indikationer
Bevacizumab i kombination med fluoropyrimidinbaseret kemoterapi er indiceret til behandling af voksne patienter med metastatisk kræft i tyktarmen og endetarmen.
Bevacizumab i kombination med paclitaxel er indiceret til førstelinjebehandling af voksne patienter med metastatisk brystkræft. Yderligere oplysninger om human epidermal vækstfaktor receptor 2 (HER2) status findes i afsnit 5.1.
Bevacizumab i kombination med capecitabin er indiceret til førstelinjebehandling af voksne patienter med metastatisk brystkræft, for hvem behandling med andre kemoterapiregimer, herunder taxaner eller antracykliner, ikke anses for hensigtsmæssig. Patienter, der har modtaget adjuverende taxan- eller anthracyclinbehandling inden for de sidste 12 måneder, bør ikke modtage behandling med Avastin i kombination med capecitabin. For mere information om HER2 -status henvises til afsnit 5.1.
Bevacizumab, som supplement til platinbaseret kemoterapi, er indiceret til førstelinjebehandling af voksne patienter med ikke-resekterbar, fremskreden, metastatisk eller tilbagevendende ikke-småcellet lungekræft med overvejende ikke-pladecellehistologi.
Bevacizumab i kombination med interferon alfa-2a er indiceret til førstelinjebehandling af voksne patienter med fremskreden og / eller metastatisk nyrecellekarcinom.
Bevacizumab i kombination med carboplatin og paclitaxel er indiceret til førstelinjebehandling af epitelial æggestokkræft, æggeledercancer eller avanceret primær peritoneal cancer (fase III B, III C og IV, ifølge International Federation of Gynecology and Obstetrics (FIGO )) hos voksne patienter.
Bevacizumab i kombination med carboplatin og gemcitabin er indiceret til behandling af voksne patienter med første tilbagefald af kræft i æggestokkene i æggestokkene, æggeledercancer eller platinfølsom primær peritoneal kræft, som ikke tidligere har modtaget behandling med bevacizumab eller andre faktorhæmmere. faktor (VEGF) eller andre midler målrettet mod VEGF -receptoren.
Bevacizumab i kombination med paclitaxel, topotecan eller pegyleret liposomal doxorubicin er indiceret til behandling af voksne patienter med recidiverende epitelial æggestokkræft, æggeledercancer eller platinresistent primær peritoneal kræft, der ikke har modtaget mere end to kemoterapibehandlinger tidligere, og som ikke har modtaget kræftbehandling tidligere behandling med bevacizumab eller andre vaskulære endotelvækstfaktor (VEGF) -hæmmere eller andre VEGF -receptor -målrettende midler (se pkt. 5.1).
Bevacizumab i kombination med paclitaxel og cisplatin eller alternativt paclitaxel og topotecan hos kvinder, der ikke kan behandles med platin, er indiceret til behandling af voksne patienter med vedvarende, tilbagevendende eller metastatisk livmoderhalskræft (se pkt.5.1).
04.2 Dosering og indgivelsesmåde
Avastin skal administreres under opsyn af en læge med erfaring i brug af antineoplastiske lægemidler.
Dosering
Metastatisk kræft i tyktarmen og endetarmen (mCRC)
Den anbefalede dosis Avastin, administreret ved intravenøs infusion, er 5 mg / kg eller 10 mg / kg legemsvægt én gang hver 2. ugeeller 7,5 mg / kg eller 15 mg / kg legemsvægt én gang hver 3. uge.
Det anbefales at fortsætte behandlingen indtil sygdomsprogression eller indtil uacceptabel toksicitet viser sig.
Metastatisk brystkræft (mBC)
Den anbefalede dosis Avastin er 10 mg / kg legemsvægt givet hver anden uge eller 15 mg / kg legemsvægt givet hver tredje uge som en intravenøs infusion.
Det anbefales at fortsætte behandlingen indtil sygdomsprogression eller indtil uacceptabel toksicitet viser sig.
Ikke-småcellet lungekræft (NSCLC)
Avastin gives som et supplement til platinbaseret kemoterapi i op til 6 behandlingscyklusser, efterfulgt af Avastin alene indtil sygdomsprogression.
Den anbefalede dosis Avastin er 7,5 mg / kg eller 15 mg / kg legemsvægt givet hver tredje uge ved intravenøs infusion.
Klinisk fordel er påvist hos NSCLC -patienter med både 7,5 mg / kg og 15 mg / kg (se pkt. 5.1).
Det anbefales at fortsætte behandlingen indtil sygdomsprogression eller indtil uacceptabel toksicitet viser sig.
Avanceret og / eller metastatisk nyrecellekarcinom (mRCC)
Den anbefalede dosis Avastin er 10 mg / kg legemsvægt, der skal administreres en gang hver anden uge ved intravenøs infusion.
Det anbefales at fortsætte behandlingen indtil sygdomsprogression eller indtil uacceptabel toksicitet viser sig.
Epitelial æggestokkræft, æggelederkræft og primær peritoneal kræft
Frontlinjebehandling: Avastin gives ud over carboplatin og paclitaxel i op til 6 behandlingscyklusser, efterfulgt af administration af Avastin alene for at fortsætte indtil sygdomsprogression eller i op til 15 måneder eller indtil uacceptabel toksicitet indtræder, alt efter hvad der indtræder først.
Den anbefalede dosis Avastin er 15 mg / kg legemsvægt, der skal administreres en gang hver 3. uge ved intravenøs infusion.
Behandling af platinfølsom sygdomstilbagefald: Avastin gives i kombination med carboplatin og gemcitabin i 6 cyklusser op til maksimalt 10 cyklusser efterfulgt af Avastin alene for at fortsætte indtil sygdomsprogression. Den anbefalede dosis Avastin er 15 mg / kg legemsvægt, der skal administreres en gang hver 3. uge ved intravenøs infusion.
Behandling af tilbagefald af platinresistent sygdom : Avastin gives i kombination med et af følgende midler: paclitaxel, topotecan (givet hver uge) eller pegyleret liposomal doxorubicin. Den anbefalede dosis Avastin er 10 mg / kg legemsvægt, der skal administreres en gang hver anden uge ved intravenøs infusion. Hvis Avastin gives i kombination med topotecan (givet på dag 1-5, hver 3. uge), er den anbefalede dosis Avastin 15 mg / kg legemsvægt, givet hver 3. uge som en intravenøs infusion. Det anbefales, at behandlingen fortsættes indtil sygdomsprogression eller udvikling af uacceptabel toksicitet (se pkt.5.1, undersøgelse MO22224).
Livmoderhalskræft
Avastin gives i kombination med en af følgende kemoterapiregimer: paclitaxel og cisplatin eller paclitaxel og topotecan.
Den anbefalede dosis Avastin er 15 mg / kg legemsvægt, der skal administreres en gang hver 3. uge ved intravenøs infusion.
Det anbefales, at behandlingen fortsættes indtil progression af den underliggende sygdom eller fremkomsten af uacceptabel toksicitet (se pkt.5.1).
Særlige patientpopulationer
Ældre patienter: Ingen dosisjustering af Avastin er påkrævet hos ældre patienter.
Patienter med nyreinsufficiens: sikkerhed og effekt hos patienter med nyreinsufficiens er ikke undersøgt (se pkt. 5.2).
Patienter med leverinsufficiens: sikkerhed og effekt hos patienter med leverinsufficiens er ikke undersøgt (se pkt. 5.2).
Pædiatrisk population
Bevacizumabs sikkerhed og effekt hos børn og unge er ikke fastslået. Der er ingen relevant anvendelse af bevacizumab hos den pædiatriske population inden for de godkendte indikationer. Aktuelt tilgængelige data er beskrevet i afsnit 5.1, 5.2 og 5.3, men ingen anbefalinger om dosering kan blive lavet.
Avastin bør ikke bruges til børn i alderen 3 til 18 år med tilbagefald eller progression af gliom af høj kvalitet på grund af effektproblemer (se pkt. 5.1 for resultaterne af undersøgelser hos pædiatriske patienter).
Dosisreduktion forbundet med bivirkninger anbefales ikke. Hvis det er angivet, skal behandlingen seponeres permanent eller midlertidigt suspenderes som beskrevet i afsnit 4.4.
Indgivelsesmåde
Startdosis bør administreres som en 90 minutters intravenøs infusion. Hvis den første infusion tolereres godt, kan den anden gives i løbet af 60 minutter. Hvis 60 minutters infusion tolereres godt, kan alle efterfølgende infusioner gives i løbet af 30 minutter.
Det må ikke administreres ved hurtig intravenøs infusion eller intravenøs bolus.
Forholdsregler, der skal træffes, før lægemidlet håndteres eller administreres
For instruktioner om fortynding af lægemidlet før administration, se afsnit 6.6. Avastin infusioner må ikke administreres eller blandes med glucoseopløsninger. Dette lægemiddel må ikke blandes med andre lægemidler end dem, der er nævnt i afsnit 6.6.
04.3 Kontraindikationer
• Overfølsomhed over for det aktive stof eller over for et eller flere af hjælpestofferne anført i pkt. 6.1.
• Overfølsomhed over for celleprodukter fra kinesisk hamster (CHO) eller andre humane eller humaniserede rekombinante antistoffer.
• Graviditet (se afsnit 4.6).
04.4 Særlige advarsler og passende forholdsregler ved brug
For at forbedre sporbarheden af biologiske lægemidler skal handelsnavnet for det administrerede produkt tydeligt registreres (eller angives) i patientens journal.
Gastrointestinale (GI) perforationer og fistler (se afsnit 4.8)
Patienter kan have en øget risiko for at udvikle gastrointestinal perforering og galdeblæreperforation under behandling med Avastin. Hos patienter med metastatisk kræft i tyktarmen eller endetarmen kan en intra-abdominal inflammatorisk proces være en risikofaktor for gastrointestinal perforering, derfor skal der udvises forsigtighed ved behandling af disse patienter. Tidligere strålebehandling er en risikofaktor for gastrointestinal perforering hos patienter behandlet med Avastin for vedvarende, tilbagevendende eller metastatisk livmoderhalskræft, og alle patienter med GI -perforeringer har tidligere været under bestråling. Hos patienter, der udvikler gastrointestinal perforering, bør behandlingen seponeres permanent.
Vagino-gastrointestinale fistler i undersøgelse GOG-0240
Patienter behandlet med Avastin for vedvarende, tilbagevendende eller metastatisk livmoderhalskræft kan have en øget risiko for at udvikle fistler mellem skeden og enhver del af mave-tarmkanalen (vagino-gastrointestinale fistler). Tidligere strålebehandling er en af de største risikofaktorer for udviklingen af vagino-gastrointestinale fistler, og alle patienter med vagino-gastrointestinale fistler har tidligere været under bestråling. Tilbagefald af carcinom i tidligere bestrålede områder er en vigtig yderligere risikofaktor for udvikling af vagino-gastrointestinale fistler.
Ikke-GI fistler (se afsnit 4.8)
Patienter kan have øget risiko for at udvikle fistler, mens de behandles med Avastin.
Hos patienter, der udvikler en tracheoesophageal (TE) fistel eller en hvilken som helst grad 4-fistel [ifølge US National Cancer Institute Common Terminology Criteria for Adverse Events (NCI-CTCAE v.3)], bør Avastin-behandlingen afbrydes. Absolut. Begrænset information er tilgængelig om fortsat brug af Avastin til patienter med andre fistler.I tilfælde af interne fistler, der ikke udvikler sig i mave -tarmkanalen, bør seponering af Avastin overvejes.
Komplikationer i helingsprocessen (se afsnit 4.8)
Avastin kan påvirke helingsprocessen negativt. Alvorlige komplikationer, herunder anastomotiske komplikationer, er blevet rapporteret i helingsprocessen med fatalt udfald. Terapi bør ikke startes i mindst 28 dage efter større operation eller indtil operationssåret er helt helet.Hos patienter, der oplever komplikationer i helingsprocessen under behandlingen, skal behandlingen afbrydes, indtil arret er helet fuldstændigt. Behandlingen skal afbrydes i tilfælde af elektiv kirurgi.
Tilfælde af nekrotiserende fasciitis, nogle dødelige, er sjældent blevet rapporteret hos patienter behandlet med Avastin. Denne tilstand er typisk forårsaget af sårhelingskomplikationer, gastrointestinale perforationer eller fisteldannelse. Hos patienter, der udvikler nekrotiserende fasciitis, bør Avastin -behandlingen afbrydes, og passende behandling indledes straks.
Forhøjet blodtryk (se afsnit 4.8)
En højere forekomst af hypertension er blevet observeret hos patienter behandlet med Avastin. Kliniske sikkerhedsdata indikerer, at forekomsten af hypertension sandsynligvis vil være dosisafhængig. Eksisterende hypertension bør kontrolleres tilstrækkeligt, inden behandling med Avastin påbegyndes. Der er ingen data om effekten af Avastin hos patienter, der har ukontrolleret hypertension på tidspunktet for behandlingens start. Blodtryksovervågning anbefales generelt under behandlingen.
I de fleste tilfælde blev hypertension kontrolleret tilstrækkeligt med standard antihypertensiv behandling, der var tilpasset den berørte patients individuelle situation.Brug af diuretika til behandling af hypertension anbefales ikke til patienter på cisplatinbaseret kemoterapiregime. Avastin bør seponeres permanent, hvis det er klinisk signifikant hypertension kan ikke kontrolleres tilstrækkeligt med antihypertensiv behandling, eller hvis patienten oplever hypertensiv krise eller hypertensiv encefalopati.
Posterior reversibel encephalopathysyndrom (PRES) (se afsnit 4.8)
Der har været sjældne rapporter om patienter behandlet med Avastin, som har oplevet tegn og symptomer relateret til PRES, en sjælden neurologisk lidelse, der blandt andet kan have følgende tegn og symptomer: anfald, hovedpine, ændret mental status, synsforstyrrelse eller kortikale blindhed, uanset om den er forbundet med hypertension eller ej. Diagnose af PRES kræver bekræftelse af hjernens radiologi, fortrinsvis magnetisk resonansbilleddannelse (MRI). Hos patienter, der oplever PRES, anbefales behandling af specifikke symptomer, herunder kontrol af hypertension og seponering af Avastin. Sikkerheden forbundet med genoptagelse af Avastin -behandling hos patienter, der tidligere har oplevet PRES, er ukendt.
Proteinuri (se afsnit 4.8)
Patienter med tidligere hypertension kan have en øget risiko for at udvikle proteinuri ved behandling med Avastin. Nogle data indikerer, at proteinuri af alle kvaliteter (ifølge US National Cancer Institute Common Terminology Criteria for Adverse Events [NCI-CTCAE v.3]) kan være dosisrelateret. Inden behandlingen påbegyndes og under den samme, anbefales det at foretage en overvågning af proteinuri ved hjælp af urinalyse med teststrimler. Hos patienter, der udvikler proteinuri af grad 4 (nefrotisk syndrom) (NCI-CTCAE v.3), bør behandlingen seponeres permanent.
Arteriel tromboemboli (se afsnit 4.8)
I kliniske forsøg var forekomsten af arterielle tromboemboliske reaktioner, herunder tilfælde af cerebralt slagtilfælde (CVA), forbigående iskæmiske anfald (TIA) og myokardieinfarkt (MI) højere hos patienter behandlet med Avastin plus kemoterapi end hos patienter i kemoterapi. alene.
Patienter behandlet med kemoterapi sammen med Avastin, som tidligere har haft arteriel tromboemboli, diabetes eller over 65 år, har en øget risiko for at udvikle arterielle tromboemboliske reaktioner under behandlingen. Der bør udvises en vis forsigtighed ved behandling af disse patienter med Avastin.
Hos patienter, der oplever arterielle tromboemboliske reaktioner, bør behandlingen seponeres permanent.
Venøs tromboemboli (se afsnit 4.8)
Patienter behandlet med Avastin kan have risiko for venøse tromboemboliske hændelser, herunder lungeemboli.
Patienter behandlet med Avastin i kombination med paclitaxel og cisplatin for vedvarende, tilbagevendende eller metastatisk livmoderhalskræft kan have øget risiko for venøse tromboemboliske hændelser.
Avastinbehandling bør afbrydes hos patienter med livstruende (grad 4) tromboemboliske reaktioner, herunder lungeemboli (NCI-CTCAE v.3). Patienter med grad ≤ 3 tromboemboliske reaktioner bør overvåges nøje (NCI-CTCAE v.3).
Blødning
Patienter behandlet med Avastin har en øget risiko for blødning, især forbundet med kræft. Avastin-behandling bør seponeres permanent hos patienter, der oplever grad 3 eller 4 blødning under Avastin-behandling (NCI-CTCAE v.3) (se pkt. 4.8).
Patienter med ubehandlede metastaser i centralnervesystemet (CNS) blev rutinemæssigt ekskluderet fra kliniske forsøg med Avastin baseret på radiologiske undersøgelser eller tegn og symptomer. Derfor er risikoen for CNS -blødning i denne patientkategori ikke blevet vurderet fremadrettet i randomiserede kliniske forsøg (se pkt. 4.8). Patienter bør overvåges for tegn og symptomer på CNS -blødning, og behandling med Avastin bør stoppes i tilfælde af intrakraniel blødning.
Der er ingen data om Avastins sikkerhedsprofil hos patienter med medfødt hæmoragisk diatese, erhvervet koagulopati eller hos patienter behandlet med fuld dosis antikoagulantia for tromboemboli før behandling med Avastin påbegyndes, da disse patienter blev udelukket fra kliniske forsøg. observeret, før behandling påbegyndes hos disse patienter.Patienter, der udvikler venøs trombose under behandlingen, ser imidlertid ikke ud til at have en øget risiko for grad 3 eller større blødning, når de behandles samtidigt med fuld dosis warfarin og Avastin (NCI-CTCAE v.3).
Lungeblødning / hæmoptyse
Patienter med ikke-småcellet lungekræft behandlet med Avastin kan have risiko for alvorlig og i nogle tilfælde dødelig lungeblødning / hæmoptyse. Patienter med nylig påbegyndt lungeblødning / hæmoptyse (> 2,5 ml knaldrødt blod) bør ikke behandles med Avastin.
Congestiv hjertesvigt (ICC) (se afsnit 4.8)
Reaktioner i overensstemmelse med en diagnose af CHF er blevet rapporteret i kliniske undersøgelser. De stødte symptomer varierede fra asymptomatisk reduktion i udkastningsfraktion i venstre ventrikel til symptomatisk CHF, der kræver behandling eller hospitalsindlæggelse. Der skal udvises forsigtighed ved behandling af patienter med klinisk signifikant hjerte-kar-sygdom, f.eks. Eksisterende koronar hjertesygdom eller CHF med Avastin.
De fleste patienter, der oplevede CHF, havde metastatisk brystkræft og tidligere havde modtaget behandling med antracykliner, strålebehandling i venstre brystvæg eller havde andre risikofaktorer for CHF.
Hos patienter fra AVF3694g -studie, der modtog behandling med antracyklin, og som ikke tidligere havde modtaget antracykliner, var der ingen stigning i forekomsten af CHF for alle grader i bevacizumab + -antracyklingruppen sammenlignet med antracykliner alene. Begyndelse af grad 3 eller højere CHF var undertiden hyppigere hos patienter behandlet med bevacizumab plus kemoterapi end hos patienter, der får kemoterapi alene. Denne observation er i overensstemmelse med resultaterne observeret hos patienter fra andre metastatisk brystkræftstudier, som ikke havde modtaget samtidig behandling med antracyklin (NCI-CTCAE v.3) (se pkt. 4.8).
Neutropeni og infektioner (se afsnit 4.8)
Hos patienter behandlet med myelotoksisk kemoterapiregimer sammen med Avastin er der observeret højere frekvenser af alvorlig neutropeni, febril neutropeni eller infektion med eller uden alvorlig neutropeni (herunder nogle med dødelig udgang) sammenlignet med kemoterapi alene. Dette er hovedsageligt blevet observeret i kombination med platin- eller taxanbaserede terapier til behandling af NSCLC, mBC og i kombination med paclitaxel og topotecan ved vedvarende, tilbagevendende eller metastatisk livmoderhalskræft.
Overfølsomhedsreaktioner / infusionsreaktioner (se afsnit 4.8)
Patienter kan have risiko for at udvikle infusions- / overfølsomhedsreaktioner. Tæt observation af patienten under og efter administration af bevacizumab anbefales som forventet for enhver humaniseret monoklonal antistofinfusion. Hvis der opstår en reaktion, skal infusionen stoppes og passende medicinsk behandling administreres Systematisk præmedicinering er ikke berettiget.
Osteonekrose i kæben (ONM) (se afsnit 4.8)
Tilfælde af ONM er blevet rapporteret hos kræftpatienter behandlet med Avastin, hvoraf de fleste tidligere eller samtidig havde modtaget intravenøs bisphosphonatbehandling, for hvilken ONM er en kendt risiko.
Der skal udvises forsigtighed, når Avastin og intravenøse bisphosphonater administreres samtidigt eller sekventielt.
Invasive tandbehandlinger er også blevet identificeret som en risikofaktor. Tandvurdering og passende tandforebyggelse bør overvejes inden behandling med Avastin. Hvis det er muligt, bør invasive tandbehandlinger undgås hos patienter, der tidligere har modtaget eller er i intravenøs bisphosphonatbehandling.
Intravitreal brug
Avastin er ikke formuleret til intravitreal brug
Øjenlidelser
Alvorlige okulære bivirkninger i både individuelle og patientgrupper er blevet rapporteret efter den ikke -godkendte intravitreale anvendelse af Avastin, bestående af hætteglas godkendt til intravenøs administration til kræftpatienter Disse reaktioner omfatter infektiøs endophthalmitis, intraokulær inflammation såsom steril endophthalmitis, uveitis, vitreitis, retinal løsrivelse, revne i nethinden pigmentepitel, øget intraokulært tryk, intraokulære blødninger såsom intravitreale blødninger eller nethindeblødninger og konjunktivalblødninger. Nogle af disse reaktioner har ført til varierende grad af synstab, herunder permanent blindhed.
Systemiske effekter efter intravitreal brug
En reduktion i cirkulerende VEGF-koncentration er blevet påvist efter intravitreal anti-VEGF-behandling. Systemiske bivirkninger såsom ikke-okulære blødninger og arterielle tromboemboliske reaktioner er blevet rapporteret efter intravitreal injektion af VEGF-hæmmere.
Ovariesvigt / fertilitet
Avastin kan forringe kvindelig fertilitet (se afsnit 4.6 og 4.8). Derfor bør terapeutiske strategier for at bevare fertiliteten diskuteres med patienter i den fertile alder, inden behandling med Avastin påbegyndes.
04.5 Interaktioner med andre lægemidler og andre former for interaktion
Virkning af antineoplastiske midler på bevacizumabs farmakokinetik
Baseret på resultaterne fra en populationsfarmakokinetisk analyse blev der ikke observeret klinisk relevante farmakokinetiske interaktioner af samtidig kemoterapi på Avastins farmakokinetik.Der var ingen statistisk signifikante eller klinisk relevante forskelle i clearance af Avastin hos patienter, der modtog Avastin. Monoterapi versus patienter. som fik Avastin i kombination med interferon alfa-2a eller andre kemoterapier (IFL, 5-FU / LV, carboplatin / paclitaxel, capecitabin, doxorubicin eller cisplatin / gemcitabin).
Bevacizumabs virkning på farmakokinetikken af andre antineoplastiske midler
Resultater fra et lægemiddelinteraktionsstudie viste ingen signifikant effekt af bevacizumab på farmakokinetikken af irinotecan og dets aktive metabolit SN38.
Resultaterne af en undersøgelse af patienter med metastatisk kolorektal cancer viste ingen signifikant effekt af bevacizumab på capecitabins farmakokinetik og dets metabolitter og på oxaliplatins farmakokinetik, bestemt af det frie og samlede platinassay.
Resultaterne af en undersøgelse af patienter med nyrecellecarcinom viste ikke nogen signifikant effekt af bevacizumab på farmakokinetikken af interferon alfa-2a.
Bevacizumabs potentielle virkning på cisplatins og gemcitabins farmakokinetik blev undersøgt hos patienter med ikke-squamøs NSCLC.Resultaterne af undersøgelsen viste, at bevacizumab ikke har nogen signifikant effekt på cisplatins farmakokinetik.Givet den høje interpatient og prøvevariation er begrænset, resultaterne af denne undersøgelse tillader ikke at drage endelige konklusioner om bevacizumabs indvirkning på gemcitabin farmakokinetik.
Kombination af bevacizumab og syg sunitinib
I to kliniske undersøgelser af metastatisk nyrecellecarcinom blev der rapporteret om mikroangiopatisk hæmolytisk anæmi (MAHA) hos 7 ud af 19 patienter, der blev behandlet med kombinationen bevacizumab (10 mg / kg hver anden uge) og sunitinibmalat (50 mg / dag).
MAHA er en hæmolytisk sygdom, der kan opstå med fragmentering af røde blodlegemer, anæmi og trombocytopeni. Derudover er hypertension (herunder hypertensive kriser), forhøjet kreatinin og neurologiske symptomer blevet observeret hos nogle af disse patienter. Alle disse manifestationer var reversible ved seponering af bevacizumab og syg sunitinib (se Hypertension, proteinuri og PRES i afsnit 4.4).
Forening med platinbaserede eller taxanbaserede terapier (se afsnit 4.4 og 4.8)
Større frekvenser af alvorlig neutropeni, febril neutropeni eller infektion med eller uden alvorlig neutropeni (herunder nogle med dødelig udgang) er hovedsageligt blevet observeret hos patienter behandlet med platin- eller taxanbaserede terapier ved behandling af NSCLC og mBC.
Strålebehandling
Sikkerheden og effekten af samtidig administration af strålebehandling og Avastin er ikke fastslået.
EGFR monoklonale antistoffer i kombination med bevacizumab-holdige kemoterapiregimer
Der er ikke udført interaktionsundersøgelser. EGFR monoklonale antistoffer bør ikke administreres til behandling af mCRC i kombination med bevacizumab-holdige kemoterapiregimer. Resultaterne af de randomiserede fase III-forsøg, PACCE og CAIRO-2, hos patienter med mCRC tyder på, at brugen af monoklonale antistoffer mod henholdsvis EGFR panitumumab og cetuximab i kombination med bevacizumab sammen med kemoterapi er forbundet med en reduktion i overlevelse. fri for progression (PFS) og / eller total overlevelse (OS) og højere toksicitet end bevacizumab sammen med kemoterapi alene.
04.6 Graviditet og amning
Kvinder i den fertile alder
Kvinder i den fertile alder bør anvende effektiv prævention under (og op til 6 måneder efter) behandling.
Graviditet
Der er ingen kliniske undersøgelsesdata fra brug af bevacizumab hos gravide Dyrestudier har vist reproduktionstoksicitet, herunder misdannelser (se pkt. 5.3). IgG vides at krydse placenta, og Avastin forventes at hæmme fostrets angiogenese og menes derfor at forårsage alvorlige medfødte abnormiteter, når det administreres under graviditet. Efter markedsføring er der observeret tilfælde af fosterabnormaliteter hos kvinder behandlet med bevacizumab som monoterapi eller i kombination med kendte embryotoksiske kemoterapeutiske midler (se pkt. 4.8) Avastin er kontraindiceret under graviditet (se pkt. 4.3).
Fodringstid
Det vides ikke, om bevacizumab udskilles i modermælk. Fordi moderens IgG udskilles i mælk og bevacizumab kan forringe barnets vækst og udvikling (se pkt. 5.3), bør kvinder stoppe amningen under behandlingen og undgå amning i mindst seks måneder derefter. Tage den sidste dosis Avastin.
Fertilitet
Toksicitetsundersøgelser ved gentagne doser hos dyr har vist, at bevacizumab kan have en negativ effekt på kvindelig fertilitet (se pkt. 5.3). I et fase III adjuverende behandlingsstudie udført hos patienter med tyktarmskræft viste en "parallel analyse hos præmenopausale patienter en" højere forekomst af nye tilfælde af ovariesvigt i bevacizumab -gruppen end i kontrolgruppen. De fleste patienter genoprettede ovariefunktionen efter afslutning af bevacizumab -behandlingen. De langsigtede virkninger af bevacizumab-behandling på fertiliteten er ukendte.
04.8 Bivirkninger
Resumé af sikkerhedsprofilen
Den overordnede sikkerhedsprofil for Avastin er baseret på data indsamlet i kliniske forsøg med over 5200 patienter med forskellige kræftformer, hovedsageligt behandlet med Avastin i kombination med kemoterapi.
De mest alvorlige bivirkninger var følgende:
• gastrointestinal perforering (se pkt. 4.4),
• blødning, herunder lungeblødning / hæmoptyse, som er mere almindelig hos patienter med ikke-småcellet lungekræft (se pkt. 4.4),
• arteriel tromboemboli (se pkt. 4.4).
De hyppigst observerede bivirkninger i kliniske undersøgelser hos patienter behandlet med Avastin var hypertension, træthed eller asteni, diarré og mavesmerter.
Analyse af kliniske sikkerhedsdata indikerer, at begyndelsen af hypertension og proteinuri forbundet med Avastin -behandling sandsynligvis vil være dosisafhængig.
Liste over bivirkninger i form af en tabel
Bivirkninger anført i dette afsnit falder ind i følgende frekvenskategorier: Meget almindelig (≥ 1/10); almindelig (≥ 1/100 y
Tabel 1 og 2 viser bivirkninger forbundet med brug af Avastin i kombination med forskellige kemoterapiregimer ved flere indikationer.
Tabel 1 viser alle bivirkninger rangeret efter hyppighed, hvis årsagssammenhæng til Avastin blev bestemt på grundlag af:
• sammenlignede forekomster identificeret mellem behandlingsgrupper i kliniske forsøg (med en forskel på mindst 10% sammenlignet med kontrolarmen for NCI-CTCAE grad 1-5 reaktioner eller en forskel på mindst 2% i forhold til kontrollen for grad 3-5 reaktioner i henhold til "NCI-CTCAE),
• sikkerhedsundersøgelser efter godkendelse
• spontan indberetning,
• epidemiologiske / ikke-interventionelle eller observationsstudier
• eller gennem en vurdering af enkeltsager.
Tabel 2 angiver hyppigheden af alvorlige bivirkninger. Alvorlige reaktioner defineres som bivirkninger med en forskel på mindst 2% fra kontrolgruppen i kliniske forsøg for grad 3-5-reaktioner i henhold til NCI-CTCAE. Tabel 2 indeholder også bivirkninger, der ifølge MA-indehaverne overvejes klinisk signifikant eller alvorlig.
Bivirkninger efter markedsføring er inkluderet i både tabel 1 og tabel 2, hvis det er relevant. Detaljeret information om disse reaktioner efter markedsføring findes i tabel 3.
Bivirkninger placeres i den passende frekvenskategori fra nedenstående tabeller baseret på den højeste forekomst, der er observeret i nogen indikation.
Inden for hver frekvenskategori præsenteres bivirkninger efter faldende sværhedsgrad.
Nogle af bivirkningerne er reaktioner, der almindeligvis ses ved kemoterapi; Avastin kan imidlertid forværre disse reaktioner, når det kombineres med kemoterapeutiske midler. Eksempler omfatter palmar-plantar erythrodysæstesi syndrom med pegyleret liposomal doxorubicin eller capecitabin, perifer sensorisk neuropati med paclitaxel eller oxaliplatin og sømforstyrrelse eller alopeci med paclitaxel
Tabel 1 Bivirkninger sorteret efter hyppighed
Når hændelser blev identificeret i kliniske forsøg som enten enhver grad eller grad 3-5 bivirkninger, blev den højeste hyppighed observeret hos patienter rapporteret. Dataene justeres ikke for forskellig behandlingstid.
a Yderligere oplysninger findes i tabel 3 "Bivirkninger rapporteret efter markedsføring".
b Termer repræsenterer en samling begivenheder, der beskriver et medicinsk begreb frem for en enkelt tilstand eller foretrukne udtryk MedDRA (Medical Dictionary for Regulatory Activities). Denne gruppe af medicinske termer kan indebære den samme underliggende patofysiologi (f.eks. Arterielle tromboemboliske reaktioner omfatter cerebrovaskulær ulykke, myokardieinfarkt, forbigående iskæmisk angreb og andre arterielle tromboemboliske reaktioner).
c Baseret på en delundersøgelse, der involverede 295 patienter fra NSABP C-08.
d Yderligere oplysninger findes i det følgende afsnit "Yderligere oplysninger om specifikke alvorlige bivirkninger".
e Rektovaginale fistler er de mest almindelige af GI -fistler.
Tabel 2 Alvorlige bivirkninger sorteret efter hyppighed
Tabel 2 angiver hyppigheden af alvorlige bivirkninger Alvorlige reaktioner defineres som bivirkninger med en forskel på mindst 2% fra kontrolarmen i kliniske forsøg med NCI-CTCAE grad 3-5-reaktioner. Tabel 2 omfatter også bivirkninger, der overvejes af Indehaveren af markedsføringstilladelsen skal være klinisk signifikant eller alvorlig. Disse klinisk signifikante bivirkninger er blevet rapporteret i kliniske undersøgelser, men grad 3-5-reaktioner nåede ikke tærsklen til en forskel på mindst 2% i forhold til kontrolgruppen. Tabel 2 omfatter også klinisk signifikante bivirkninger observeret kun efter markedsføringen, derfor er frekvensen og graden ifølge NCI-CTCAE ikke kendt. Derfor er disse klinisk signifikante reaktioner inkluderet i tabel 2 i kolonnen med titlen "Frekvens ikke kendt".
a Vilkår repræsenterer en samling begivenheder, der beskriver et medicinsk begreb frem for en enkelt tilstand eller foretrukne udtryk MedDRA (Medical Dictionary for Regulatory Activities). Denne gruppe af medicinske termer kan indebære den samme underliggende patofysiologi (f.eks.arterielle tromboemboliske reaktioner omfatter cerebrovaskulær ulykke, myokardieinfarkt, forbigående iskæmisk anfald og andre arterielle tromboemboliske reaktioner).
b For yderligere information henvises til det følgende afsnit "Yderligere oplysninger om specifikke alvorlige bivirkninger".
c Yderligere oplysninger findes i tabel 3 "Bivirkninger rapporteret efter markedsføring"
d Rektovaginale fistler er de mest almindelige af GI-vaginale fistler.
Beskrivelse af specifikke alvorlige bivirkninger
Gastrointestinale (GI) perforationer og fistler (se afsnit 4.4)
Avastinbehandling har været forbundet med alvorlige episoder af gastrointestinal perforering.
Gastrointestinale perforationer er blevet rapporteret i kliniske forsøg med en "forekomst på mindre end 1% hos patienter med metastatisk brystkræft eller med ikke-småcellet og ikke-pladelig lungekræft, op til 2,0% hos patienter med metastatisk nyrekræft eller hos patienter med æggestokkræftpatienter, der er i behandling i første række og op til 2,7% (inklusive gastrointestinal fistel og byld) hos patienter med metastatisk kolorektal cancer. I et klinisk studie af patienter med vedvarende, tilbagevendende eller metastatisk livmoderhalskræft (undersøgelse GOG-0240) blev der rapporteret GI-perforeringer (af alle kvaliteter) hos 3,2% af patienterne, som alle tidligere havde gennemgået bækkenbestråling.
Typen og sværhedsgraden, hvormed disse hændelser opstod, var forskellige: fra tilstedeværelsen af fri luft detekteret ved direkte abdominal radiografi, som forsvandt uden behandling, til intestinal perforering med abdominal abscess og fatalt udfald. I nogle tilfælde var der underliggende abdominal betændelse på grund af mavesår, tumornekrose, diverticulitis eller kemoterapi-associeret colitis.
Omkring en tredjedel af de alvorlige tilfælde af gastrointestinal perforering var dødelig. Dette tal repræsenterer 0,2% -1% af alle patienter behandlet med Avastin.
Gastrointestinale fistler (af enhver klasse) er blevet rapporteret i kliniske forsøg med Avastin med en "maksimal forekomst på 2% hos patienter med kræft i æggestokkene og metastatisk kolorektal cancer. Sådanne fistler blev imidlertid mindre almindeligt rapporteret hos patienter med andre kræftformer.
Vagino-gastrointestinale fistler i undersøgelse GOG-0240
I en undersøgelse foretaget hos patienter med vedvarende, tilbagevendende eller metastatisk livmoderhalskræft var forekomsten af GI-fistler 8,3% hos patienter behandlet med Avastin og 0,9% hos patienter i kontrolarmen., Alle tidligere underkastet bækkenbestråling Hyppigheden af vagino-gastrointestinal fistler i Avastin + kemoterapigruppen var højere hos patienter med tilbagefald i tidligere bestrålede områder (16,7%) end hos patienter med tilbagefald i områder, der ikke blev bestrålet. til tidligere bestråling (3,6%). Tilsvarende frekvenser i kontrolgruppen kun for kemoterapi var 1,1% vs. 0,8% Patienter, der udvikler GI-vaginale fistler, kan også opleve tarmobstruktion og kræve operation og stomipakning.
Ikke-GI fistler (se afsnit 4.4)
Avastinbehandling har været forbundet med alvorlige episoder med fistler, hvoraf nogle var dødelige.
I en klinisk undersøgelse af patienter med vedvarende, tilbagevendende eller metastatisk livmoderhalskræft (GOG-240) blev der rapporteret om ikke-gastrointestinale fistler, der påvirker kvindelig vagina, blære eller kønsorganer hos 1,8% af patienterne behandlet med Avastin. Og hos 1,4% af patienterne i kontrolarmen.
Ikke almindelige manifestationer (≥ 0,1% - galde) er blevet observeret i de forskellige indikationer. Fistler er også blevet rapporteret efter markedsføring.
Reaktioner er blevet rapporteret på forskellige tidspunkter under behandlingen, der spænder fra en uge til mere end 1 år efter behandlingens start med Avastin, hvor de fleste reaktioner forekom inden for de første 6 måneder af behandlingen.
Helbredelsesproces (se afsnit 4.4)
Fordi Avastin -behandling kan påvirke helingsprocessen negativt, blev patienter, der havde gennemgået en større operation inden for de foregående 28 dage, udelukket fra fase III -undersøgelserne.
I kliniske forsøg med metastatisk kræft i tyktarmen eller endetarmen var der ingen tegn på øget risiko for postoperativ blødning eller komplikationer i helingsprocessen hos patienter, der gennemgik en større operation 28 til 60 dage før initiering af Avastin -behandling. En "øget forekomst af postoperativ blødning eller komplikationer i helingsprocessen, der forekom inden for 60 dage efter større kirurgi, blev observeret hos patienter behandlet med Avastin på operationstidspunktet. Incidensen varierede mellem 10% (4/40) og 20% (3/15).
Der er rapporteret om alvorlige sårhelingskomplikationer, herunder anastomotiske komplikationer, hvoraf nogle har været dødelige.
I metastatiske eller lokalt tilbagevendende brystkræftstudier blev grad 3-5 helbredelseskomplikationer observeret hos op til 1,1% af patienterne behandlet med Avastin sammenlignet med op til 0,9% af patienterne i kontrolarmene (NCI-CTCAE v.3).
I kliniske forsøg med ovariecancer blev grad 3-5 sårhelingskomplikationer observeret hos op til 1,2% af patienterne i bevacizumab-armen vs. 0,1% af kontrolarmen (NCI-CTCAE v.3).
Forhøjet blodtryk (se afsnit 4.4)
En højere forekomst af hypertension (alle kvaliteter) på op til 42,1% blev observeret hos patienter behandlet med Avastin i kliniske undersøgelser sammenlignet med 14% hos dem, der blev behandlet med kontrol. Grad 3 og 4 hypertension (der kræver orale antihypertensive lægemidler) blev observeret hos 0,4% -17,9% af patienterne behandlet med Avastin. Grad 4 hypertension (hypertensiv krise) forekom hos 1,0% af patienterne behandlet med Avastin og kemoterapi sammenlignet med 0,2% af patienterne behandlet med den samme kemoterapi alene (NCI-CTCAE v.3).
Generelt er hypertension blevet kontrolleret tilstrækkeligt med orale antihypertensiva, såsom angiotensinkonverterende enzymhæmmere, diuretika og calciumkanalblokkere. Denne hændelse har sjældent resulteret i afbrydelse af Avastin -behandling eller hospitalsindlæggelse.
Meget sjældne tilfælde af hypertensiv encefalopati er blevet rapporteret, hvoraf nogle har været dødelige.
Risikoen for hypertension forbundet med Avastin -behandling var ikke relateret til patienternes baseline -karakteristika, underliggende sygdom eller samtidige behandlinger.
Posterior reversibel encephalopathysyndrom "." (PRES) (se afsnit 4.4)
I sjældne tilfælde er der rapporteret tegn og symptomer relateret til PRES, en sjælden neurologisk lidelse, under behandlingen af patienter med Avastin. Manifestationer kan omfatte anfald, hovedpine, ændret mental status, synsforstyrrelser eller kortikal blindhed, med eller uden tilhørende hypertension. Den kliniske præsentation af PRES er ofte uspecifik, så diagnosen PRES kræver bekræftelse ved hjernebilleddannelse, fortrinsvis magnetisk resonansbilleddannelse (MRI).
Hos patienter, der er mistænkt for PRES, anbefales tidlig genkendelse af specifikke symptomer og deres behandling, herunder kontrol af hypertension (hvis det er forbundet med alvorlig ukontrolleret hypertension), ud over at afslutte behandling med bevacizumab. Symptomerne forsvinder eller forbedres normalt inden for få dage efter behandlingen er stoppet, selvom nogle patienter har oplevet nogle neurologiske følger. Sikkerheden forbundet med genstart af Avastin -behandling hos patienter, der tidligere har oplevet PRES, er ukendt.
Otte tilfælde af PRES blev rapporteret i alle kliniske forsøg. To ud af otte tilfælde havde ikke radiologisk bekræftelse af MR.
Proteinuri (se afsnit 4.4)
I kliniske undersøgelser blev proteinuri fundet hos mellem 0,7% og 38% af patienterne behandlet med Avastin.
Proteinuri manifesterede sig med en sværhedsgrad, der varierede fra et klinisk asymptomatisk, forbigående og sporbart proteinuri til et nefrotisk syndrom; i de fleste tilfælde var det proteinuri af grad 1 (NCI-CTCAE v.3). Grad 3 proteinuri blev rapporteret hos op til 8,1% af de behandlede patienter. Grad 4 proteinuri (nefrotisk syndrom) blev observeret hos 1,4% af de behandlede patienter. Proteinuri observeret i kliniske forsøg med Avastin var ikke forbundet med nyresvigt og krævede sjældent permanent seponering af behandlingen.Det anbefales, at proteinuri kontrolleres, inden Avastin -behandling påbegyndes.I mange kliniske undersøgelser førte proteinuri -niveauer ≥ 2 g / 24 timer til seponering af Avastin, indtil niveauet faldt til under 2 g / 24 timer.
Blødning (se afsnit 4.4)
I kliniske forsøg med alle indikationer varierede den samlede forekomst af NCI-CTCAE v.3 grad 3-5 blødningsreaktioner fra 0,4% til 6,9% hos patienter behandlet med Avastin sammenlignet med maksimalt 4,5% af patienterne i kontrolgruppen med kemoterapi.
I et klinisk studie med patienter med vedvarende, tilbagevendende eller metastatisk livmoderhalskræft (undersøgelse GOG-0240) blev der rapporteret grad 3-5 hæmoragiske reaktioner hos op til 8,3% af patienterne, der blev behandlet med Avastin i kombination med paclitaxel. Og topotecan versus maksimalt 4,6% af patienter behandlet med paclitaxel og topotecan.
De hæmoragiske reaktioner, der blev observeret i kliniske forsøg, var overvejende tumorassocierede blødninger (se nedenfor) og mindre slimhindeblødninger (f.eks. Epistaxis).
Tumorassocieret blødning (se afsnit 4.4)
Massiv eller større lungeblødning / hæmoptyse er hovedsageligt blevet observeret i undersøgelser hos patienter med ikke-småcellet lungekræft (NSCLC). Mulige risikofaktorer omfatter: pladecelleshistologi, behandling med antirheumatiske / antiinflammatoriske lægemidler, behandling med antikoagulantia, tidligere strålebehandling, Avastin-behandling, aterosklerose, central tumorplacering og tumorkavitation før eller under behandlingen. De eneste variabler, der viste statistisk signifikante korrelationer med blødning, var Avastin -behandling og pladecellehistologi.NCCLC -patienter med bekræftet pladecelle- eller blandet pladecellehistologi blev ekskluderet fra efterfølgende faseundersøgelser. III, mens patienter med ukendt tumorhistologi var inkluderet.
Hos patienter med NSCLC, med udelukkelse af dem med overvejende pladecelleshistologi, blev reaktioner af alle grader påvist med en frekvens på op til 9% ved behandling med Avastin og kemoterapi sammenlignet med 5% hos patienter behandlet med kemoterapi alene. Der blev observeret 3-5 reaktioner hos op til 2,3% af patienterne behandlet med Avastin og kemoterapi sammenlignet med
Gastrointestinale blødninger, herunder rektal blødning og melaena er blevet rapporteret hos patienter med tyktarmskræft og er blevet vurderet som tumorassocierede blødninger.
Tumorassocieret blødning er også sjældent blevet rapporteret på andre typer og steder af tumorer, herunder tilfælde af centralblødning (CNS) hos patienter med CNS-metastaser (se pkt. 4.4).
Forekomsten af CNS-blødninger hos patienter med ikke-forbehandlede CNS-metastaser, der modtog bevacizumab, er ikke blevet prospektivt vurderet i randomiserede kliniske forsøg. Kræft, 3 ud af 91 (3,3%) patienter med hjernemetastaser havde CNS-blødninger (alle grader 4), når de blev behandlet med bevacizumab , sammenlignet med 1 tilfælde (grad 5) af 96 patienter (1%), som ikke blev udsat for bevacizumab. I to efterfølgende undersøgelser af patienter med forbehandlede hjernemetastaser (involverende ca. 800 patienter) forekom et tilfælde af grad 2 CNS -blødning hos 83 patienter behandlet med bevacizumab (1,2%) på analysetidspunktet. midlertidig (NCI-CTCAE v.3).
I alle kliniske forsøg med Avastin blev der observeret "slimhindeblødning" hos op til 50% af patienterne, der blev behandlet med Avastin. De fleste af disse var NCI-CTCAE v.3 grad 1 næseblod. Varede mindre end 5 minutter og forsvandt uden medicinsk intervention og uden skal ændre Avastin doseringsplan. Kliniske sikkerhedsdata tyder på, at forekomsten af mindre slimhindeblødning (f.eks. Epistaxis) kan være dosisafhængig.
Mindre slimhindeblødningsreaktioner på andre steder blev også registreret sjældnere; for eksempel tandkødsblødning eller vaginal blødning.
Tromboemboli (se afsnit 4.4)
Arteriel tromboemboli: En "øget forekomst af arterielle tromboemboliske reaktioner, herunder cerebrovaskulære ulykker, myokardieinfarkt, forbigående iskæmiske anfald og andre arterielle tromboemboliske reaktioner blev observeret hos patienter behandlet med Avastin i alle indikationer.
I kliniske forsøg var den samlede forekomst af arterielle tromboemboliske reaktioner op til 3,8% i de Avastin-holdige arme sammenlignet med op til 1,7% i kemoterapikontrolarmene. Dødelige hændelser blev rapporteret hos 0,8% af patienterne behandlet med Avastin sammenlignet med 0,5% af patienterne behandlet med kemoterapi alene. Cerebrovaskulære ulykker (inklusive forbigående iskæmiske angreb) blev rapporteret hos op til 2,3% af patienterne, der blev behandlet med Avastin i kombination med kemoterapi, sammenlignet med 0,5% af patienterne, der blev behandlet med kemoterapi alene. Myokardieinfarkt blev registreret hos 1,4% af patienterne behandlet med Avastin i kombination med kemoterapi sammenlignet med 0,7% af patienterne behandlet med kemoterapi alene.
I et klinisk studie, der evaluerede Avastin i kombination med 5-fluorouracil / folinsyre, AVF2192g, blev patienter med metastatisk kolorektal cancer, som ikke var kandidater til behandling med irinotecan, inkluderet. I dette studie blev arterielle tromboemboliske reaktioner observeret hos 11% (11/100) af patienterne sammenlignet med 5,8% (6/104) i kontrolgruppen med kemoterapi.
Venøs tromboemboli: I kliniske forsøg var forekomsten af venøs tromboemboliske reaktioner ens hos patienter behandlet med Avastin i kombination med kemoterapi sammenlignet med dem, der blev behandlet med kontrolkemoterapi alene. Venøse tromboemboliske reaktioner omfatter dyb venetrombose, lungeemboli og tromboflebitis.
I kliniske undersøgelser for alle indikationer varierede den samlede forekomst af venøse tromboemboliske reaktioner fra 2,8% til 17,3% af patienterne behandlet med Avastin sammenlignet med 3,2% -15,6% i kontrolgrupperne.
Grad 3-5 venøse tromboemboliske reaktioner (NCI-CTCAE v.3) er blevet rapporteret hos op til 7,8% af patienterne behandlet med kemoterapi plus bevacizumab sammenlignet med op til 4,9% af patienterne, der kun blev behandlet med kemoterapi (i de forskellige indikationer, med undtagelse af vedvarende, tilbagevendende eller metastatisk livmoderhalskræft).
I en klinisk undersøgelse af patienter med vedvarende, tilbagevendende eller metastatisk livmoderhalskræft (undersøgelse GOG-0240) blev grad 3-5 venøse tromboemboliske hændelser rapporteret hos op til 15,6% af patienterne behandlet med Avastin i kombination med paclitaxel. Og cisplatin mod maksimum 7,0 % af patienterne behandlet med paclitaxel og cisplatin.
Patienter, der har oplevet en venøs tromboembolisk reaktion, kan have større risiko for tilbagefald, hvis de får Avastin i kombination med kemoterapi end kemoterapi alene.
Congestiv hjertesvigt (ICC):
I kliniske forsøg med Avastin forekom kongestiv hjertesvigt (CHF) i alle kræftindikationer, der er undersøgt til dato, men forekom hovedsageligt hos patienter med metastatisk brystkræft.I de fire fase III -undersøgelser (AVF2119g, E2100, BO17708 og AVF3694g) hos patienter med metastatisk brystkræft grad 3 (NCI-CTCAE v.3) eller højere er blevet rapporteret med en "forekomst på op til 3,5% af patienterne behandlet med Avastin i kombination med kemoterapi sammenlignet med et maksimum på 0, 9% i kontrolgrupperne. For patienter inkluderet i studiet AVF3694g behandlet med anthracycliner samtidigt med bevacizumab var forekomsten af CHF grad 3 eller højere for de respektive bevacizumab- og kontrolarme den, der blev observeret i andre metastatiske brystkræftstudier: 2,9% i armen anthracyclin + bevacizumab og 0 %i anthracyclin + placebo -armen. I undersøgelse AVF3694g var den observerede forekomst af enhver CHF -grad ens for anthracyclin + Avastin -armen (6, 2%) og for anthracyclin + placebo -armen (6,0%).
De fleste patienter, der udviklede CHF under mBC kliniske forsøg, viste forbedring i venstre ventrikels symptomer og / eller funktion efter passende medicinsk behandling.
I de fleste kliniske forsøg med Avastin, patienter med allerede eksisterende NYHA fase II-IV CHF (New York Heart Association) blev udelukket, og der er derfor ingen information tilgængelig om risikoen for CHF i denne population.
Tidligere eksponering for anthracykliner og / eller tidligere strålebehandling af brystvæggen kan repræsentere risikofaktorer for udvikling af CHF.
En øget forekomst af CHF blev observeret i et klinisk studie med patienter med diffust stort B -cellelymfom ved behandling med bevacizumab i kombination med en kumulativ dosis doxorubicin større end 300 mg / m2.Denne fase III-undersøgelse havde til formål at sammenligne rituximab / cyclophosphamid / doxorubicin / vincristine / prednison (R-CHOP) i kombination med bevacizumab med R-CHOP uden bevacizumab. Højere, end det, der tidligere blev observeret for doxorubin, var procentdelen højere i den behandlede arm med R-CHOP og bevacizumab. Disse resultater tyder på, at omhyggelig klinisk observation med passende kardiologisk vurdering bør overvejes hos patienter udsat for kumulative doser af doxorubicin større end 300 mg / m2 i kombination med bevacizumab.
Overfølsomhedsreaktioner / infusionsreaktioner (se afsnit 4.4 og Efter markedsføring erfaring under)
Hyppigere anafylaktiske eller anafylaktoide reaktioner er blevet rapporteret i nogle kliniske forsøg med patienter, der får Avastin i kombination med kemoterapi end hos dem, der får kemoterapi alene. Forekomsten af disse reaktioner i nogle kliniske forsøg med Avastin er almindelig (op til 5% af patienterne behandlet med bevacizumab).
Infektioner
I et klinisk studie med patienter med vedvarende, tilbagevendende eller metastatisk livmoderhalskræft (undersøgelse GOG-0240) blev grad 3-5-infektioner rapporteret hos op til 24% af patienterne behandlet med Avastin i kombination med paclitaxel og topotecan mod maksimum 13% af patienter behandlet med paclitaxel og topotecan.
Ovariesvigt / fertilitet (se afsnit 4.4 og 4.6)
I fase III NSABP C-08-studiet med Avastin i adjuverende behandling hos tyktarmskræftpatienter er forekomsten af nye tilfælde af ovariesvigt, defineret som amenoré, der varer 3 måneder eller længere, med FSH-niveauer i blodet ≥ 30 mIU / ml og negativ for serum β-HCG graviditetstest, blev analyseret hos 295 præmenopausale kvinder.Nye tilfælde af ovariesvigt blev rapporteret hos 2,6% af patienterne behandlet med mFOLFOX-6 sammenlignet med 39% i gruppen af patienter behandlet med mFOLFOX-6 + bevacizumab. afslutningen af behandlingen med bevacizumab, genoprettede ovariefunktionen hos 86,2% af de evaluerede patienter. De langsigtede virkninger af bevacizumab på fertiliteten er ukendte.
Ændringer af laboratorieparametre
Behandling med Avastin kan være forbundet med nedsat antal neutrofile og hvide blodlegemer og tilstedeværelsen af protein i urinen.
I alle kliniske undersøgelser forekom følgende grad 3 og 4 (NCI-CTCAE v.3) ændringer i laboratorieparametre hos patienter behandlet med Avastin med mindst 2% forskel fra de tilsvarende kontrolgrupper: hyperglykæmi, nedsat hæmoglobin, hypokaliæmi, hyponatriæmi , nedsat antal hvide blodlegemer, øget internationalt normaliseret forhold (INR).
Andre særlige populationer
Ældre patienter
I randomiserede kliniske forsøg har en alder> 65 år været forbundet med en øget risiko for at udvikle arterielle tromboemboliske reaktioner, herunder cerebrovaskulære ulykker (ACV), forbigående iskæmiske anfald (TIA) og myokardieinfarkt (MI). Andre reaktioner set med højere frekvens i patienter i alderen> 65 år var leukopeni og trombocytopeni i grad 3-4, neutropeni, diarré, kvalme, hovedpine og træthed af enhver grad sammenlignet med behandlede patienter i alderen ≤ 65 år med Avastin (se pkt. 4.4 og 4.8 under overskriften Tromboemboli). I et klinisk studie var forekomsten af grad ≥ 3 hypertension dobbelt så høj hos patienter i alderen> 65 år som i den yngre aldersgruppe (
Hos ældre patienter (> 65 år) behandlet med Avastin var der ingen højere forekomst af andre reaktioner, herunder gastrointestinal perforering, komplikationer i helingsprocessen, CHF og blødning sammenlignet med patienter i alderen ≤ 65 år behandlet med Avastin.
Pædiatrisk population
Avastins sikkerhed hos børn og unge er ikke fastslået.
Efter markedsføring erfaring
Tabel 3 Bivirkninger rapporteret efter markedsføring
* hvis angivet, blev hyppigheden afledt af data fra kliniske forsøg
Indberetning af formodede bivirkninger
Rapportering af formodede bivirkninger, der opstår efter godkendelse af lægemidlet, er vigtig, da det muliggør kontinuerlig overvågning af lægemidlets fordel / risiko -balance. Sundhedspersonale anmodes om at rapportere alle formodede bivirkninger via det nationale rapporteringssystem. "Adresse http: //www.agenziafarmaco.gov.it/it/responsabili.
04.9 Overdosering
Den højeste dosis, der er vurderet til mennesker (20 mg / kg legemsvægt, intravenøst hver anden uge) har været forbundet med alvorlig migræne hos mange patienter.
05.0 FARMAKOLOGISKE EGENSKABER
05.1 Farmakodynamiske egenskaber
Farmakoterapeutisk klassifikation: antineoplastiske og immunmodulerende midler, antineoplastiske midler, andre antineoplastiske midler, monoklonale antistoffer, ATC -kode: L01XC07.
Handlingsmekanisme
Bevacizumab forhindrer ved binding til vaskulær endotelcellevækstfaktor (VEGF), en vigtig promotor for vaskulogenese og "angiogenese, sidstnævnte fra at binde sig til dets receptorer, Flt-1 (VEGFR-1) og KDR (VEGFR-2), den overfladen af endotelcellerne. Blokeringen af den biologiske aktivitet af VEGF regresserer vaskularisering af tumorer, normaliserer den resterende tumorvaskularisering og hæmmer dannelsen af ny vaskularisering og forhindrer dermed tumorvækst.
Farmakodynamiske virkninger
Administration af bevacizumab eller dets tilsvarende museantistof i tumor -xenograft -modeller i nøgenmus viste "omfattende antitumoraktivitet i humane kræftformer, herunder tyktarm, bryst, bugspytkirtel og prostata. Progressionen af metastatisk sygdom blev blokeret og reduceret mikrovaskulær permeabilitet.
Klinisk effekt
Metastatisk kræft i tyktarmen eller endetarmen (mCRC)
Sikkerheden og effekten af den anbefalede dosis (5 mg / kg legemsvægt hver anden uge) ved metastatisk kræft i tyktarmen eller endetarmen blev undersøgt i tre randomiserede, aktivt kontrollerede kliniske forsøg i kombination med førstelinjes kemoterapi og en fluoropyrimidinbase Avastin blev kombineret med to kemoterapiregimer:
• Undersøg AVF2107g: Ugentlig administration af irinotecan / bolus af 5-fluorouracil / folinsyre (IFL) i i alt 4 uger af hver 6-ugers cyklus (Saltz-regime).
• Undersøg AVF0780g: i kombination med bolus 5-fluorouracil / folinsyre (5-FU / FA) i alt 6 uger af hver 8-ugers cyklus (Roswell Park-regime).
• Undersøg AVF2192g: i kombination med bolus 5-FU / FA i alt 6 uger af hver 8-ugers cyklus (Roswell Park-regime) hos patienter, der ikke anses for at være optimale kandidater til førstelinjebehandling med irinotecan.
Tre yderligere undersøgelser blev udført med bevacizumab hos patienter med mCRC: førstelinje (NO16966), andenlinie hos patienter, der ikke tidligere havde modtaget behandling med bevacizumab (E3200) og andenlinje hos patienter, der tidligere blev behandlet med førstelinje bevacizumab, som var gået frem (ML18147). I disse undersøgelser blev bevacizumab administreret i kombination med FOLFOX-4 (5FU / LV / oxaliplatin), XELOX (capecitabin / oxaliplatin) og fluoropyrimidin / irinotecan eller fluoropyrimidin / oxaliplatin i henhold til følgende doseringsregimer:
• NO16966: Avastin 7,5 mg / kg legemsvægt hver 3. uge i kombination med oral capecitabin og intravenøs oxaliplatin (XELOX) eller Avastin 5 mg / kg hver 2. uge i kombination med leucovorin plus bolus 5 -fluorouracil, efterfulgt af 5 -fluorouracil som en infusion, med intravenøs oxaliplatin (FOLFOX-4).
• E3200: Avastin 10 mg / kg legemsvægt hver 2. uge i kombination med leucovorin og bolus 5-fluorouracil, efterfulgt af infusion af 5-fluorouracil, med intravenøs oxaliplatin (FOLFOX-4) hos patienter, der ikke tidligere er blevet behandlet med bevacizumab.
ML18147: Avastin 5,0 mg / kg legemsvægt hver 2. uge eller Avastin 7,5 mg / kg legemsvægt hver 3. uge i kombination med fluoropyrimidin / irinotecan eller fluoropyrimidin / oxaliplatin hos patienter med sygdomsprogression efter behandling med første linje med bevacizumab. Brugen af et regime indeholdende irinotecan eller oxaliplatin blev ændret afhængigt af førstelinjebrug af oxaliplatin eller irinotecan.
AVF2107g
Dette randomiserede, fase III, dobbeltblinde, aktivt kontrollerede kliniske forsøg evaluerede kombinationen af Avastin med IFL i førstelinjebehandlingen af metastatisk kræft i tyktarmen eller endetarmen. Otte hundrede og tretten patienter blev randomiseret til at modtage IFL. + Placebo (arm 1) eller IFL + Avastin (5 mg / kg hver 2. uge, arm 2). En tredje gruppe på 110 patienter modtog bolus 5-FU / FA + Avastin (arm 3). Tilmelding til arm 3 blev afbrudt som forventet , når Avastins sikkerhed i kombination med IFL -regimet blev fastslået og anset for acceptabel. Alle behandlinger blev fortsat indtil sygdomsprogression. Den samlede gennemsnitsalder var 59,4 år; 56,6% af patienterne havde en præstationsstatus ECOG på 0, 43% havde et niveau på 1 og 0,4% et niveau på 2. 15,5% havde tidligere gennemgået strålebehandling og 28,4% havde kemoterapi.
Samlet overlevelse var det primære mål for evaluering af effekt i undersøgelsen. Tilføjelse af Avastin til IFL-regimet resulterede i statistisk signifikante stigninger i den samlede overlevelse, progressionsfri overlevelse og den samlede responsrate (se tabel 4.) Klinisk fordel, målt ved samlet overlevelse, blev observeret i alle undergrupper. Af præspecificerede patienter , herunder dem, der er defineret efter alder, køn, præstationsstatus, placeringen af den primære tumor, antallet af involverede organer og varigheden af metastatisk sygdom.
Effektresultaterne af Avastin i kombination med IFL -kemoterapi er vist i tabel 4.
Tabel 4 Effektresultater fra undersøgelse AVF2107g
til 5 mg / kg hver 2. uge
b I forhold til kontrolarmen
Blandt de 110 patienter, der blev randomiseret til Arm 3 (5-FU / FA + Avastin) før afbrydelse af denne arm, var medianvarigheden af den samlede overlevelse 18,3 måneder, og den mediane progressionsfri overlevelse var 8. 8 måneder.
AVF2192g
Denne randomiserede, fase II, dobbeltblinde, aktivt kontrollerede kliniske undersøgelse evaluerede effektiviteten og sikkerheden af Avastin i kombination med 5-FU / folinsyre ved førstelinjebehandling af metastatisk kolorektal cancer hos ikke-patienter. Betragtes som optimale kandidater til irinotecan førstelinjebehandling. Hundrede og fem patienter blev randomiseret til 5-FU / FA + placebo-armen og 104 patienter til 5-FU / FA + Avastin-armen (5 mg / kg hver 2. uge). Alle behandlinger blev fortsat tilføjelse til Avastin 5 mg / kg hver anden uge til 5-FU / FA resulterede i højere objektive responsrater, signifikant længere progressionsfri overlevelse og tendens til længere overlevelse end 5-FU / FA kemoterapi alene.
AVF0780g
Dette randomiserede, fase II, aktivt kontrollerede, åbne kliniske forsøg evaluerede Avastin i kombination med 5-FU / FA i førstelinjebehandlingen af metastatisk kolorektal cancer. Medianalderen var 64 år. 19% af patienterne havde tidligere modtaget kemoterapi og 14% havde strålebehandling. 71 patienter blev randomiseret til at modtage enten 5-FU / FA i bolus eller 5-FU / FA kombinationen. + Avastin (5 mg / kg hver 2. uge). En tredje gruppe på 33 patienter modtog bolus 5-FU / FA + Avastin (10 mg / kg hver 2. uge) .Patienter blev behandlet indtil sygdomsprogression. endepunkt undersøgelsens primære fokus var objektiv responsrate og progressionsfri overlevelse. Tilføjelse af Avastin 5 mg / kg hver anden uge til 5-FU / FA resulterede i højere objektive responsrater, længere progressionsfri overlevelse og en tendens til længere overlevelse sammenlignet med 5-FU / FA kemoterapi alene (se tabel 5) Denne effekt data er i overensstemmelse med resultaterne i undersøgelsen AVF2107g.
Effektdata fra undersøgelser AVF0780g og AVF2192g, som vurderede brugen af Avastin i kombination med 5-FU / FA kemoterapi, er opsummeret i tabel 5.
04.7 Virkninger på evnen til at føre motorkøretøj og betjene maskiner
Avastin har ingen eller ubetydelig indflydelse på evnen til at føre motorkøretøj eller betjene maskiner. Søvnighed og synkope er imidlertid blevet rapporteret ved brug af Avastin (se tabel 1, afsnit 4.8). Patienter, der oplever symptomer, der påvirker deres syn eller koncentration, eller deres evne til at reagere, bør rådes til ikke at køre bil og ikke bruge maskiner, før symptomer forsvinder.
Tabel 5 Effektdata fra undersøgelser AVF0780g og AVF2192g
NO16966
Dette var en fase III, randomiseret, dobbeltblind (for bevacizumab) undersøgelse, der evaluerede Avastin 7,5 mg / kg i kombination med oral capecitabin og i.v. oxaliplatin. (XELOX), givet i 3-ugers cyklusser eller Avastin 5 mg / kg i kombination med leucovorin og bolus 5-fluorouracil, efterfulgt af infusion af 5-fluorouracil, med oxaliplatin i.v. (FOLFOX-4) givet i 2-ugers cyklusser. Undersøgelsen bestod af to faser: en indledende del med 2 arme (del I), hvor patienter blev randomiseret til to forskellige behandlingsgrupper (XELOX og FOLFOX-4), og en efterfølgende del med 4 arme 2 x 2 faktoriel (del II), hvor patienter blev randomiseret til fire behandlingsgrupper (XELOX + placebo, FOLFOX-4 + placebo, XELOX + Avastin, FOLFOX-4 + Avastin). I del II blev behandlingsopgaven dobbeltblindet med hensyn til Avastin-administration.
Cirka 350 patienter blev randomiseret til hver af de 4 undersøgelsesarme i del II af undersøgelsen.
Tabel 6 Behandlingsregimer i studie NO16966 (mCRC)
Den primære parameter til vurdering af undersøgelsens effektivitet var varigheden af progressionsfri overlevelse I denne undersøgelse var der to forskellige primære mål: at demonstrere, at XELOX ikke var ringere end FOLFOX-4 og at vise, at Avastin i kombination med FOLFOX- kemoterapi. 4 eller XELOX var bedre end kemoterapi alene. Begge primære mål blev opnået:
• Ikke-mindreværd for de XELOX-holdige arme sammenlignet med FOLFOX-4-holdige arme i den samlede sammenligning blev påvist med hensyn til progressionsfri overlevelse og samlet overlevelse i den berettigede befolkning behandlet i henhold til protokol.
• De Avastin-holdige arms overlegenhed i forhold til de kemoterapiske arme i den samlede sammenligning blev påvist med hensyn til progressionsfri overlevelse i ITT-populationen (tabel 7).
Sekundære analyser af PFS, baseret på vurderingen af patienternes respons under behandling, bekræftede den signifikant overlegne kliniske fordel for patienter behandlet med Avastin (analyser vist i tabel 7), i overensstemmelse med den statistisk signifikante fordel, der blev observeret i den samlede analyse.
Tabel 7 Vigtige effektresultater for "superioritetsanalyse (ITT -population". "Undersøgelse NO16966)
* Samlet overlevelsesanalyse ved klinisk cut-off pr. 31. januar 2007
** Primær analyse ved klinisk cut-off pr. 31. januar 2006
a I forhold til kontrolarmen
I den FOLFOX-behandlede undergruppe var median PFS 8,6 måneder hos placebobehandlede patienter og 9,4 måneder hos bevacizumabbehandlede patienter, HR = 0,89, 97,5% CI = [0,73; 1,08]; p-værdi = 0,1871, mens de tilsvarende resultater i den XELOX-behandlede undergruppe var 7,4 vs. 9,3 måneder, HR = 0,77, 97,5% CI = [0,63; 0,94]; p-værdi = 0,0026.
I FOLFOX-undergruppen var den mediane samlede overlevelse 20,3 måneder hos placebobehandlede patienter og 21,2 måneder hos bevacizumabbehandlede patienter, HR = 0,94, 97,5% CI = [0,75; 1.16]; p-værdi = 0,4937, mens de tilsvarende resultater i den XELOX-behandlede undergruppe var 19,2 vs. 21,4 måneder, HR = 0,84, 97,5% CI = [0,68; 1,04]; p-værdi = 0,0698.
ECOG E3200
Dette var et randomiseret, åbent, kontrolleret fase III-studie, der evaluerede Avastin 10 mg / kg i kombination med leucovorin og bolus 5-fluorouracil efterfulgt af infusion af 5-fluorouracil med i.v. oxaliplatin. (FOLFOX-4), givet i 2-ugers cyklusser hos tidligere behandlede patienter (anden linje) med fremskreden kolorektal cancer. I kemoterapi-armene blev FOLFOX-4-regimet brugt ved de samme doser og tidsplan vist i tabel 6 til undersøgelse NO16966.
Undersøgelsens primære effektparameter var samlet overlevelse defineret som tiden fra randomisering til død af enhver årsag. Otte hundrede og niogtyve patienter blev randomiseret (292 FOLFOX-4, 293 Avastin + FOLFOX-4 og 244 Avastin) alene. .) Tilsætning af Avastin til FOLFOX-4-regimen forlængede statistisk signifikant overlevelse. Statistisk signifikante forbedringer i progressionsfri overlevelse og objektiv responsrate blev også observeret (se tabel 8).
Tabel 8 Effektresultater for undersøgelse E3200
Der var ingen signifikant forskel i den samlede overlevelsesvarighed mellem patienter, der fik Avastin monoterapi og patienter behandlet med FOLFOX-4. Progressionsfri overlevelse og objektiv respons var lavere i Avastin monoterapirammen end i FOLFOX-4-armen.
ML18147
Denne randomiserede, kontrollerede, åbne fase III kliniske undersøgelse evaluerede brugen af Avastin 5,0 mg / kg hver 2. uge eller 7,5 mg / kg hver 3. uge i kombination med fluoropyrimidin-baseret kemoterapi kontra kemoterapi. Med fluoropyrimidin monoterapi hos patienter med mCRC, der er udviklet sig efter førstelinjebehandling indeholdende bevacizumab.
Patienter med histologisk bekræftet mCRC og sygdomsprogression blev randomiseret 1: 1 inden for 3 måneder efter seponering af førstelinjebehandling med bevacizumab for at modtage fluoropyrimidin / oxaliplatin eller fluoropyrimidin / irinotecan-baseret kemoterapi (kemoterapi ændret baseret på førstelinjes kemoterapi modtaget) med eller uden kemoterapi Bevacizumab. Behandlingen blev fortsat indtil sygdomsprogression eller udvikling af uacceptabel toksicitet. Undersøgelsens primære endepunkt var samlet overlevelse defineret som tiden siden randomisering til død af enhver årsag.
820 patienter blev randomiseret. Tilføjelsen af bevacizumab til fluoropyrimidinbaseret kemoterapi resulterede i en statistisk signifikant forlængelse af overlevelsen for patienter med mCRC, der udviklede sig efter førstelinjebehandling indeholdende bevacizumab (ITT = 819) (se tabel 9).
Tabel 9 Effektresultater for undersøgelse ML18147 (ITT -population)
en 5,0 mg / kg hver 2. uge eller 7,5 mg / kg hver 3. uge
Statistisk signifikante forbedringer blev også observeret i progressionsfri overlevelse. Den objektive svarprocent var lav i begge behandlingsgrupper, og forskellen var ikke signifikant.
Undersøgelse E3200 brugte en bevacizumab -dosis på 5 mg / kg / uge til patienter, der ikke tidligere blev behandlet med bevacizumab, mens undersøgelse ML18147 brugte en bevacizumab -dosis på 2,5 mg / kg / uge til bevacizumab -forbehandlede patienter. En sammenligning mellem undersøgelser med hensyn til effekt og sikkerhed er begrænset af forskellene mellem selve undersøgelserne, især med hensyn til patientpopulation, der tidligere var behandlet med bevacizumab og kemoterapiregimer. Både bevacizumab -doser på 5 mg / kg / uge og 2,5 mg / kg / uge gav statistisk signifikant OS -fordel (HR 0,751 i studie E3200; HR 0,81 i studie ML18147) og PFS (HR 0,518 i studie E3200; HR 0,68 i studie ML18147) . Med hensyn til sikkerhed var der en højere samlet forekomst af grad 3-5 AE'er i studie E3200 sammenlignet med undersøgelse ML18147.
Metastatisk brystkræft (mBC)
To store fase III -undersøgelser blev udført med det formål at evaluere effekten af behandling med Avastin i kombination med to forskellige kemoterapiregimer med hensyn til PFS som primært endepunkt.I begge undersøgelser blev der observeret en signifikant stigning i PFS. Begge fra en klinisk og statistisk synspunkt.
PFS -resultaterne for de enkelte kemoterapeutiske midler inkluderet i indikationen er opsummeret nedenfor:
• Undersøg E2100 (paclitaxel)
• 5,6 måneders stigning i median PFS, HR 0,421 (s
• Undersøg AVF3694g (capecitabin)
• 2,9 måneders stigning i median PFS, HR 0,69 (p = 0,0002, 95% CI 0,56, 0,84)
Yderligere detaljer vedrørende hver undersøgelse findes nedenfor.
ECOG E2100
Studie E2100 er et multicenter, åbent, randomiseret, aktivt kontrolleret klinisk forsøg, der evaluerer Avastin i kombination med paclitaxel for metastatisk eller lokalt tilbagevendende brystkræft hos patienter, der ikke tidligere har modtaget kemoterapi for metastatisk sygdom og lokalt tilbagevendende. Patienter blev randomiseret til paclitaxel alene (90 mg / m2 som en 1 times intravenøs infusion en gang ugentligt i tre uger ud af fire) eller i kombination med Avastin (10 mg / kg som en IV -infusion hver anden uge). Tidligere hormonbehandling var tilladt til behandling af metastatisk sygdom. Adjuverende behandling med taxan var kun tilladt, hvis den var afsluttet mindst 12 måneder før inklusion i undersøgelsen.Blandt de 722 patienter i undersøgelsen havde størstedelen af patienterne HER2-negativ sygdom (90%), hvor et lille antal patienter enten havde ukendt (8%) eller bekræftet positiv (2%) HER2-status, der tidligere var behandlet med trastuzumab eller troede for ikke at være berettiget til trastuzumab -behandling. Desuden havde 65% af patienterne tidligere modtaget adjuvant kemoterapi, taxanbaseret i 19% af tilfældene og anthracykliner i 49% af tilfældene. Patienter med metastaser i centralnervesystemet, herunder tidligere behandlede eller resekterede hjerneskader, blev ekskluderet.
I studie E2100 blev patienter behandlet indtil sygdomsprogression. I tilfælde, der kræver tidligt ophør med kemoterapi, fortsatte behandlingen med Avastin monoterapi indtil sygdomsprogression. Patientkarakteristika var ens mellem undersøgelsens to arme. Undersøgelsens primære endepunkt var progressionsfri overlevelse (PFS), baseret på undersøgelsesundersøgernes vurdering af sygdomsprogression Derudover blev der også foretaget en uafhængig vurdering af det primære endepunkt. Resultaterne af denne undersøgelse er rapporteret i tabel 10.
Tabel 10 Effektresultater fra undersøgelse E2100
* primær analyse
Den kliniske fordel ved Avastin vurderet med hensyn til PFS blev observeret i alle foruddefinerede undergrupper analyseret (inklusive sygdomsfrit interval, antal metastatiske steder, tidligere adjuverende kemoterapiindtag og østrogenreceptorstatus (RE)).
AVF3694g
AVF3694g er et fase III, multicenter, randomiseret, placebokontrolleret studie designet til at evaluere effektiviteten og sikkerheden af Avastin i kombination med kemoterapi versus kemoterapi plus placebo i førstelinjebehandling af patienter med metastatisk eller HER2-negativ brystkræft. Lokalt tilbagevendende.
Kemoterapiregimet blev valgt efter undersøgelsens skøn inden randomisering i et forhold på 2: 1 til at modtage kemoterapi plus Avastin eller kemoterapi plus placebo. Kemoterapimuligheder inkluderede capecitabin, taxaner (proteinbundet paclitaxel, docetaxel) og anthracyclinholdige regimer (doxorubicin / cyclophosphamid, epirubicin / cyclophosphamid, 5-fluorouracil / doxorubicin / cyclophosphamid, 5-fluorouracil / epirubicin / cyclophosphamid) administreret hver 3. uge. Avastin eller placebo blev givet i en dosis på 15 mg / kg hver tredje uge.
Denne undersøgelse omfattede en blindet behandlingsfase, en valgfri åben fase efter sygdomsprogression og en opfølgningat vurdere overlevelse. I den blindede behandlingsfase modtog patienter kemoterapibehandling og medicin (Avastin eller placebo) hver 3. uge indtil sygdomsprogression, behandlingsbegrænsende toksicitet eller død. Ved dokumenteret sygdomsprogression kunne patienter, der blev placeret i den valgfrie åbne fase, modtage åben Avastin i kombination med en lang række godkendte andenlinjemedicin.
Statistiske analyser blev udført uafhængigt af de to patientkohorter: 1) patienter, der modtog capecitabin i kombination med Avastin eller placebo; 2) patienter, der gennemgår taxan- eller anthracyclinbaserede behandlinger i kombination med Avastin eller placebo. L "endepunkt det primære studie var PFS som vurderet af efterforskeren. Desuden er "endepunkt primær blev også evalueret af et uafhængigt revisionsudvalg (IRC).
Resultaterne af denne undersøgelse fra den endelige analyse defineret i protokollen og udført i den statistisk uafhængige potenskohort af patienter behandlet med capecitabin fra studie AVF3694g for progressionsfri overlevelse og responsrater er vist i tabel 11. Resultater fra en "eksplorativ samlet overlevelse analyse, der indeholder yderligere 7 måneders opfølgning (ca. 46% af patienterne var døde) er også angivet. Procentdelen af patienter, der fik Avastin i den åbne fase, var 62,1% i capecitabin + placebo-armen og 49,9% i capecitabin + Avastin-armen.
Tabel 11 Effektresultater for undersøgelse AVF3694g: "." Capecitabinaae Avastin / Placebo (Cap + Avastin / Pl)
1000 mg / m2 oralt to gange dagligt i 14 dage givet hver 3. uge
b Stratificeret analyse inklusive alle progression- og dødshændelser eksklusive dem, for hvilke ikke-protokolbehandling (NPT) blev påbegyndt forud for dokumenteret progression; data fra disse patienter blev censureret ved den sidste tumorvurdering inden starten af NPT.
En "ikke-stratificeret analyse af PFS (undersøgt vurderet) blev udført uden at censurere patienter, for hvem ikke-protokolbehandling (NPT) blev påbegyndt før sygdomsprogression. Resultaterne af disse analyser lignede meget resultaterne af den primære analyse. af PFS.
Ikke-småcellet lungekræft (NSCLC)
Sikkerheden og effekten af Avastin som supplement til platinbaseret kemoterapi ved førstelinjebehandling af patienter med ikke-squamøs ikke-småcellet lungekræft (NSCLC) blev undersøgt i undersøgelser E4599 og BO17704. Undersøgelse E4599 viste en samlet overlevelsesfordel med bevacizumab dosis på 15 mg / kg en gang hver 3. uge Undersøgelse BO17704 viste, at både bevacizumab doser på 7,5 mg / kg og 15 mg / kg en gang hver 3. uge øger progressionsfri overlevelse og responsrate.
E4599
Undersøgelse E4599 var et multicenter, åbent, randomiseret, aktivt lægemiddelkontrolleret klinisk forsøg, der evaluerede Avastin som førstelinjebehandling af patienter med lokalt avanceret NSCLC (stadium IIIb med malign pleural effusion) metastatisk eller tilbagevendende med ikke-histologi. celle.
Patienter blev randomiseret til behandling med platinbaseret kemoterapi (paclitaxel 200 mg / m2 og carboplatin AUC = 6,0, begge ved IV-infusion (PC) på dag 1 i hver cyklus fra 3 uger op til 6 cyklusser eller PC i kombination med Avastin ved en dosis på 15 mg / kg IV infusion på infusionsdag 1 i hver 3-ugers cyklus. Efter afslutning af 6 cyklusser med carboplatin-paclitaxel kemoterapi eller tidlig afbrydelse af kemoterapi, fortsatte patienterne i Avastin + carboplatin arm -paclitaxel med at modtage Avastin som monoterapi hver 3. uge indtil sygdomsprogression blev 878 patienter randomiseret i de to arme.
Under undersøgelsen modtog 32,2% (136/422) af patienterne, der modtog studiebehandling, 7-12 administrationer af Avastin og 21,1% (89/422) 13 eller flere administrationer af Avastin.
Det primære endepunkt var varigheden af overlevelse Resultaterne er vist i tabel 12.
Tabel 12 Effektresultater fra undersøgelse E4599
I en undersøgende analyse var den generelle overlevelsesfordel ved Avastin mindre relevant i undergruppen af patienter, der ikke havde adenocarcinoma histologi.
BO17704
Undersøgelse BO17704 var et randomiseret, dobbeltblindet fase III-studie af Avastin ud over cisplatin og gemcitabin versus placebo, cisplatin og gemcitabin hos patienter med lokalt avanceret ikke-squamøs NSCLC (fase III b med supraclavikulær lymfeknude metastase eller malign effusion pleural eller perikardial), metastatisk eller tilbagevendende, som ikke havde modtaget tidligere kemoterapi. L "endepunkt primær var progressionsfri overlevelse; blandt endepunkt Sekundære undersøgelser omfattede den samlede overlevelsesvarighed.
Patienter blev randomiseret til platinbaseret kemoterapi, cisplatin 80 mg / m2 intravenøs infusion på dag 1 og gemcitabin 1250 mg / m2 intravenøs infusion på dag 1 og 8 i hver 3-ugers cyklus op til 6 cyklusser (CG) med placebo eller CG med Avastin i en dosis på 7,5 eller 15 mg / kg ved IV infusion på dag 1 i hver 3-ugers cyklus. I Avastin -armene kunne patienter modtage Avastin som monoterapi hver 3. uge indtil sygdomsprogression eller utålelig toksicitet. Undersøgelsesresultaterne viste, at 94% (277/296) af de berettigede patienter fortsat modtog bevacizumab som monoterapi i cyklus 7.En høj procentdel af patienterne (ca. 62%) gennemgik talrige, uspecificerede behandlinger mod kræft, hvilket kan have haft indflydelse på den samlede overlevelsesanalyse.
Effektresultaterne er vist i tabel 13.
Tabel 13 Effektresultater fra undersøgelse BO17704
patienter med målbar sygdom ved baseline
Avanceret og / eller metastatisk nyrecellekarcinom (mRCC)
Avastin i kombination med interferon alfa-2a til førstelinjebehandling af fremskreden og / eller metastatisk nyrecellekarcinom (BO17705)
Dette var et dobbeltblindet, randomiseret fase III-studie for at evaluere effektiviteten og sikkerheden af Avastin i kombination med interferon (IFN) alfa-2a versus interferon (IFN) alfa-2a alene i førstelinjebehandling. De 649 randomiserede patienter (641 behandlet) havde en Karnofsky Performance Status (KPS) ≥ 70%, ingen CNS -metastaser og "tilstrækkelig organfunktion." Patienterne blev nefrektomiseret for primær nyrecellekarcinom. Avastin blev administreret i en dosis på 10 mg / kg hver 2. uge indtil sygdom IFN alfa-2a blev administreret i op til 52 uger eller indtil sygdomsprogression ved den anbefalede startdosis på 9 MIU tre gange om ugen, hvilket muliggjorde en dosisreduktion op til 3 MIU tre gange om ugen i 2 faser. Patienter blev stratificeret efter land og Motzer -kriterier og behandlingsarme var godt afbalanceret med hensyn til prognostiske faktorer det.
Slutpunktet primære i undersøgelsen var samlet overlevelse, med endepunkt sekundær inklusive progressionsfri overlevelse. Tilføjelsen af Avastin til IFN-alpha-2a øgede signifikant PFS og den objektive responsrate.Disse resultater blev bekræftet af en uafhængig radiologisk gennemgang.To måneders stigning iendepunkt primær overlevelse var ikke signifikant (HR = 0,91). En stor andel af patienterne (ca. 63% IFN / placebo; 55% Avastin / IFN) modtog en række uspecificerede kræftbehandlinger, herunder antineoplastiske midler, der kan have påvirket den samlede overlevelsesvurdering, efter undersøgelsesafslutning.
Effektresultaterne er vist i tabel 14.
Tabel 14 Effektresultater fra undersøgelse BO17705
En "multivariat undersøgende analyse ifølge Cox -regressionsmodellen ved hjælp af foruddefinerede parametre, indikerede, at følgende prognostiske faktorer, der blev vurderet ved baseline, var tæt korreleret med overlevelse, uanset behandling: køn, hvide blodlegemer og trombocyttal, fald i kropsvægt i de 6 måneder før tilmelding, antal metastatiske steder, summen af større diametre af mållæsioner, Motzer -kriterier. Justering for disse faktorer resulterede i et hazard ratio på 0,78 (95% CI [0,63; 0,96], p = 0,0219), hvilket indikerer en reduktion på 22% i dødsrisikoen for patienter i Avastin + IFN alfa-2a-armen i forhold til dem i IFN alfa-2a-armen.
Syvoghalvfems patienter i IFN alfa-2a-armen og 131 patienter i Avastin-armen reducerede dosis IFN alfa 2a fra 9 MIU til 6 eller 3 MIU tre gange om ugen som specificeret i protokollen. Dosisreduktionen af IFN alfa-2a ser ikke ud til at have påvirket effekten af kombinationen af Avastin og IFN alfa-2a med hensyn til PFS, hvilket fremgår af en undergruppeanalyse. De 131 patienter i Avastin + IFN behandlingsarmen alfa- 2a, der reducerede og fastholdt dosen af IFN alfa-2a til 6 eller 3 MIU under undersøgelsen, havde en 6, 12 og 18 måneders sygdomsfri overlevelsesrate på henholdsvis 73, 52 og 21%sammenlignet med 61, 43 og 17% i den globale population af patienter behandlet med Avastin og IFN alfa-2a.
AVF2938
Dette var et randomiseret, dobbeltblindet, fase II klinisk forsøg med at studere Avastin 10 mg / kg i en 2-ugers tidsplan versus Avastin i samme dosis i kombination med erlotinib 150 mg dagligt hos patienter med nyrecellecarcinom. Klarcellet metastatisk . I denne undersøgelse blev i alt 104 patienter randomiseret til behandling, 53 med Avastin 10 mg / kg hver 2. uge plus placebo og 51 med Avastin 10 mg / kg hver anden uge plus erlotinib 150 mg dagligt. "Analysen af"endepunkt primær adskilte sig ikke mellem Avastin + placebo -armen og Avastin + erlotinib -armen (median PFS 8,5 versus 9,9 måneder). Syv patienter i hver arm havde et objektivt svar. Tilsætning af erlotinib til bevacizumab resulterede ikke i forbedring i OS (HR 1.764; p = 0.1789), varighed af objektiv respons (6,7 versus 9,1 måneder) eller tid til symptomprogression (HR = 1,172; p = 0,5076).
AVF0890
Dette var et randomiseret fase II -studie for at sammenligne effektiviteten og sikkerheden af bevacizumab versus placebo. I alt 116 patienter blev randomiseret til at modtage bevacizumab med 3 mg / kg hver 2. uge (n = 39), 10 mg / kg hver 2. uge ( n = 37) eller placebo (n = 40) midlertidig viste, at der var en signifikant forlængelse af tiden til sygdomsprogression i gruppen på 10 mg / kg sammenlignet med placebogruppen (hazard ratio 2,55; p
Epitel æggestokkene, æggelederen og primær peritoneal kræft
Førstelinjebehandling af kræft i æggestokkene
Sikkerheden og effekten af Avastin ved førstelinjebehandling af patienter med epitelial æggestokkene, æggeleder eller primær peritoneal cancer blev evalueret i to fase III kliniske forsøg (GOG-0218 og BO17707), der vurderede virkningerne af "Tilføjelse af Avastin til en carboplatin og paclitaxel -behandling kontra kemoterapi alene.
GOG-0218
Undersøgelse GOG-0218 var et fase III, multicenter, randomiseret, dobbeltblindet, placebokontrolleret trearmet studie, der evaluerede effekten af at tilføje Avastin til et godkendt kemoterapiregime (carboplatin og paclitaxel) hos patienter med epitelial æggestokkræft, æggeleder rørkræft eller fremskreden primær peritoneal kræft (FIGO fase IIIB, IIIC og IV).
Patienter, der tidligere blev behandlet for kræft i æggestokkene med bevacizumab eller antineoplastisk behandling (f.eks. Kemoterapi, monoklonal antistofbehandling, tyrosinkinasehæmmerbehandling eller hormonbehandling) eller patienter, der tidligere havde modtaget strålebehandling af maven eller maven, blev ekskluderet fra undersøgelsen.
I alt 1873 patienter blev randomiseret i samme forhold i følgende tre arme:
• CPP -arm: Fem placebo -cykler (initieret fra cyklus 2) i kombination med carboplatin (AUC 6) og paclitaxel (175 mg / m2) i 6 cykler efterfulgt af administration af placebo kun i op til 15 måneders behandling
• CPB15 -arm: Fem cyklusser Avastin (15 mg / kg q3w startet fra 2. cyklus) i kombination med carboplatin (AUC 6) og paclitaxel (175 mg / m2) i 6 cykler efterfulgt af administration af placebo kun i op til 15 måneder af terapi
• CPB15 + arm: Fem kurser Avastin (15 mg / kg q3w startet fra 2. cyklus) i kombination med carboplatin (AUC 6) og paclitaxel (175 mg / m2) i 6 cykler efterfulgt af kontinuerlig administration af Avastin monoterapi (15 mg / kg q3w) op til 15 måneders behandling.
De fleste af patienterne i undersøgelsen var hvide (87% på tværs af de tre arme); medianalderen var 60 år i CPP og CPB15 armen og 59 år i CPB15 + armen; 29% af patienterne i CPP og CPB15 og 26% i CPB15 + armen var ældre end 65 år. Ca. 50% af alle patienter havde et GOG PS på 0 ved baseline, cirka 43% et GOG PS på 1 og cirka 7% et GOG PS på 2. De fleste patienter havde en EOC -diagnose (82% i CPP og CPB15, 85 % i CPB15 +), PPC (16% i CPP, 15% i CPB15, 13% i CPB15 +) og FTC (1% i CPP, 3% i CPB15, 2% i CPB15 +). De fleste patienter havde serøs type adenocarcinom (85% i CPP og CPB15, 86% i CPB15 +).Ca. 34% af alle tilmeldte patienter var i FIGO trin III optimalt resekteret med evaluerbar restsygdom, 40% i FIGO trin III med suboptimal radikalisering, og 26% var i FIGO trin IV.
Det primære endepunkt blev undersøgt-vurderet PFS i betragtning af sygdomsprogression baseret på radiologisk billeddannelse, CA 125-niveauer eller forværring af symptomer som defineret i protokollen. Desuden blev der foretaget en på forhånd specificeret dataanalyse ved at censurere for progressionshændelser defineret på basis af CA 125 værdier samt en uafhængig vurdering af PFS som funktion af radiologiske vurderinger.
Undersøgelsen opfyldte det primære mål om forbedring af PFS. Sammenlignet med patienter behandlet med kemoterapi alene (carboplatin og paclitaxel) i førstelinjebehandlingen, patienter, der fik bevacizumab i en dosis på 15 mg / kg q3w i kombination med kemoterapi, og som fortsatte at modtage bevacizumab monoterapi (CPB15 +), påvist klinisk og statistisk signifikant forbedring af PFS.
Hos patienter behandlet med bevacizumab alene i kombination med kemoterapi, og som ikke fortsatte med monoterapi med bevacizumab (CPB15), blev der ikke observeret nogen klinisk signifikant forbedring af PFS.
Resultaterne af denne undersøgelse er opsummeret i tabel 15.
Tabel 15 Effektresultater fra undersøgelse GOG-0218
1 Undersøger-vurderet PFS-analyse baseret på protokolspecificerede GOG-parametre (patienter, der hverken er censureret for CA 125-defineret progression eller NPT før sygdomsprogression) med dataafbrydelse pr. 25. februar 2010.
2 Sammenlignet med kontrolarmen; stratificeret fareforhold.
3 Ensidig log-rank test, p-værdi
4 Værdi af s grænse lig med 0,0116.
5 Patienter med evaluerbar sygdom ved baseline.
6 Endelig samlet overlevelsesanalyse udført, når 46,9% af patienterne var døde.
Forud specificerede analyser af PFS blev udført, alle med en skæringsdato den 29. september 2009. Resultaterne af disse analyser er som følger:
• Den efterforsker-vurderede PFS-analyse specificeret i protokollen (ikke censureret for progression defineret af tumormarkør CA 125 og NPT-værdier) viste et stratificeret fareforhold på 0,71 (95% CI: 0, 61-0,83; 1-sidet log rank test , p-værdi
• Den primære analyse af PFS vurderet af efterforskerne (censurering efter anden progression defineret af CA-125-værdierne og for NPT) viste et stratificeret fareforhold på 0,62 (95% CI: 0,52- 0,75, 1-sidet log rangtest, p-værdi
• PFS-analyse bestemt af det uafhængige revisionsudvalg (censurering for NPT) viste et stratificeret fareforhold på 0,62 (95% CI: 0,50-0,77, ensidig lograngstest, p-værdi
PFS -analyser efter undergrupper relateret til sygdomsstadium og primær kirurgi er vist i tabel 16. Disse resultater bekræfter robustheden af PFS -analysen som vist i tabel 15.
Tabel 16 - Resultater af PFS1 efter sygdomsstadie og kirurgi som følge af undersøgelse GOG -0218
1 Undersøger-vurderet PFS-analyse baseret på protokolspecificerede GOG-parametre (patienter, der hverken er censureret for CA 125 defineret progression eller NPT før sygdomsprogression) med dataafbrydelse pr. 25. februar 2010.
2 Med grov restsygdom
3 3,7% af alle randomiserede patienter var i fase IIIB sygdom
4Relativ til kontrolarmen
BO17707 (ICON7)
BO17707 er et fase III, to-armet, multicenter, randomiseret, kontrolleret, åbent studie med det formål at evaluere effekten af at tilføje Avastin til carboplatin og paclitaxel efter operation hos patienter med epitelial æggestokkræft. Æggelederkarcinom eller primært peritonealt karcinom stadium I eller IIA i henhold til FIGO -klassificeringen (grad 3 eller klar cellehistologisk undertype; n = 142) eller fase IIB - IV i henhold til FIGO -klassificeringen (alle grader og alle typer histologiske, n = 1386) (NCI -CTCAE v.3) .
Patienter, der tidligere var blevet behandlet med bevacizumab eller antineoplastisk behandling af kræft i æggestokkene (f.eks. Kemoterapi, monoklonal antistofbehandling, tyrosinkinasehæmmerbehandling eller hormonbehandling) eller patienter, der tidligere havde modtaget strålebehandling, blev ekskluderet fra undersøgelsen.
I alt 1528 patienter blev randomiseret i samme forhold i følgende to arme:
• CP -arm: Carboplatin (AUC 6) og paclitaxel (175 mg / m2) i 6 cyklusser, der varer 3 uger
• CPB7.5 + arm: Carboplatin (AUC 6) og paclitaxel (175 mg / m2) i 6 cyklusser hver 3. uge i kombination med Avastin (7,5 mg / kg q3w) i op til 12 måneder (administration af Avastin startes fra den 2. kemoterapicyklus, hvis behandlingen blev startet inden for 4 uger efter operationen eller fra 1. cyklus, hvis behandlingen blev startet mere end 4 uger efter operationen).
De fleste af patienterne i undersøgelsen var hvide kaukasiske (96%), medianalderen var 57 år i begge behandlingsgrupper, 25% af patienterne var 65 år eller ældre og cirka 50% af patienterne havde et PS på 1 på ECOG -skala og 7%af patienterne i hver behandlingsarm havde en ECOG PS på 2. De fleste patienter havde diagnosen EOC (87,7%) efterfulgt af PPC (6,9%) og FTC (3,7%) eller en "blandet histologi (1,7%) De fleste patienter var FIGO trin III (68% i begge) efterfulgt af fase IV i henhold til FIGO -klassifikationen (13% og 14%), fase II ifølge FIGO -klassificeringen (10% og 11%) og fase I i henhold til FIGO -klassificeringen (9% og 7%). de fleste patienter i hver behandlingsarm (74% og 71%) havde en indledende diagnose af dårligt differentieret neoplasma (grad 3). Forekomsten af histolundertyper EOC -symptomer var ens på tværs af behandlingsarme; 69% af patienterne i hver arm havde adenocarcinom af serøs type.
L "endepunkt primær var PFS, vurderet af undersøgeren ved hjælp af RECIST.
Undersøgelsen opfyldte sit primære mål med hensyn til PFS-forbedring. Sammenlignet med patienter, der blev behandlet med førstelinjens kemoterapi alene (carboplatin og paclitaxel), patienter, der fik bevacizumab i en dosis på 7,5 mg / kg q3w i kombination med kemoterapi, og som fortsatte med at tage bevacizumab i op til 18 cyklusser viste statistisk signifikant forbedring af PFS.
Resultaterne af denne undersøgelse er opsummeret i tabel 17.
Tabel 17 Effektresultater fra undersøgelse BO17707 (ICON7)
1 Hos patienter med målbar sygdom ved baseline.
2 Undersøger-vurderet PFS-analyse med dataafbrydelse pr. 30. november 2010.
3 Den endelige analyse af den samlede overlevelse blev udført med dataafbrydelsen pr. 31. marts 2013, da 46,7% af patienterne var døde.
Den primære efterforsker-vurderede PFS-analyse med dataforkortelse, der dateres tilbage til 28. februar 2010, viste et ustratificeret fareforhold på 0,79 (95% CI: 0,68-0,91, log-rank test på 2-sidet, p-værdi 0,0010) med en median PFS på 16,0 måneder i CP -armen og 18,3 måneder i CPB7,5 + -armen.
PFS -analysen af undergrupper relateret til sygdomsstadium og primær kirurgi er vist i tabel 18. Disse resultater bekræfter robustheden af PFS -analysen som rapporteret i tabel 17.
Tabel 18 - PFS1 -resultater efter sygdomsstadie og kirurgi fra undersøgelse BO17707 (ICON7)
1 Undersøger-vurderet PFS-analyse med dataforkortelse pr. 30. november 2010.
2 Med eller uden grov restsygdom
3 5,8% af alle patienter var fase IIIB -sygdom
4Relativ til kontrolarmen
Tilbagevendende kræft i æggestokkene
Sikkerheden og effekten af Avastin til behandling af tilbagevendende epitelial æggestokkræft, æggeledercancer eller primær peritoneal cancer blev undersøgt i to fase III -undersøgelser (AVF4095g og MO22224) med forskellige kemoterapiregimer og patientpopulationer.
• Undersøgelse AVF4095g evaluerede effektiviteten og sikkerheden af bevacizumab i kombination med carboplatin og gemcitabin hos patienter med tilbagevendende platinfølsom epitelial æggestok, æggeleder eller primær peritoneal cancer.
• Undersøgelse MO22224 evaluerede effektiviteten og sikkerheden af bevacizumab i kombination med paclitaxel, topotecan eller pegyleret liposomal doxorubicin hos patienter med recidiverende epitelial æggestokkene, æggeleder eller platinresistent primær peritoneal cancer.
AVF4095g
Den randomiserede fase III, dobbeltblindede, placebokontrollerede undersøgelse (AVF4095g) evaluerede sikkerheden og effekten af Avastin til behandling af patienter med tilbagefald af platinfølsom sygdom ved epitelial æggestokkræft, æggelederkræft eller kræft. Peritoneal sygdom, som ikke tidligere havde modtaget kemoterapi for tilbagefald eller tidligere behandling med bevacizumab. Undersøgelsen sammenlignede effekten af at tilføje Avastin til carboplatin og gemcitabin kemoterapi efterfulgt af fortsat brug af Avastin alene indtil sygdommens progression, sammenlignet med kemoterapi med carboplatin og gemcitabin alene.
Kun patienter med æggestokkræft, primær peritoneal kræft eller histologisk dokumenteret æggelederkræft, der kom tilbage mindst 6 måneder efter afslutning af platinbaseret kemoterapi, og som hverken havde modtaget tilbagefald af kemoterapi eller tidligere behandling, blev inkluderet i undersøgelsen. Med bevacizumab eller anden VEGF hæmmere eller andre VEGF -receptor -målrettende midler.
I alt 484 patienter med målbar sygdom blev randomiseret 1: 1 til:
• carboplatin (AUC4, dag 1) og gemcitabin (1000 mg / m2 på dag 1 og 8) og samtidig placebo hver 3. uge i 6 cykler og op til 10 cykler efterfulgt af placebo alene (hver 3. uge) indtil sygdomsprogression eller uacceptabel toksicitet .
• carboplatin (AUC4, dag 1) og gemcitabin (1000 mg / m2 på dag 1 og 8) og samtidig Avastin (15 mg / kg på dag 1) hver 3. uge i 6 cyklusser og op til 10 cykler efterfulgt af Avastin (15 mg / kg hver 3. uge) alene indtil sygdomsprogression eller uacceptabel toksicitet.
L "endepunkt primær var progressionsfri overlevelse baseret på undersøgelsesvurdering med modificeret RECIST 1.0. Mereendepunkt de omfattede objektiv respons, responsvarighed, samlet overlevelse og sikkerhed. En uafhængig gennemgang af " endepunkt primær.
Resultaterne af denne undersøgelse er opsummeret i tabel 19.
Tabel 19 Effektresultater fra undersøgelse AVF4095
PFS -analyser for undergrupper defineret af tid til tilbagefald fra den sidste platineterapi er opsummeret i tabel 20.
Tabel 20 Progressionsfri overlevelse fra sidste platineterapi til tilbagefald
MO22224
Undersøgelse MO22224 evaluerede effektiviteten og sikkerheden af bevacizumab i kombination med kemoterapi til behandling af tilbagevendende kræft i æggestokkene i æggestokkene, æggelederkræft eller platinresistent primær peritoneal cancer. Forsøgsdesignet omfattede et fase III-studie. etiket til vurdering af bevacizumab -behandling i kombination med kemoterapi (CT + BV) versus kemoterapi alene (CT).
Undersøgelsen omfattede i alt 361 patienter, der modtog kemoterapi alene [paclitaxel, topotecan eller pegyleret liposomal doxorubicin (PLD)] eller kemoterapi i kombination med bevacizumab:
• CT -arm (kun kemoterapi):
• paclitaxel 80 mg / m2 som en i.v. infusion 1 time på dag 1, 8, 15 og 22 hver 4. uge.
• topotecan 4 mg / m2 som en i.v. 30 minutter på dag 1, 8 og 15 hver 4. uge. Alternativt kan en dosis på 1,25 mg / m2 administreres i 30 minutter på dag 1-5 hver 3. uge.
• PLD 40 mg / m2 som en i.v. infusion 1 mg / min på dag 1 kun hver 4. uge. Efter cyklus 1 kan medicinen administreres som en 1 times infusion.
• CT + BV -arm (kemoterapi + bevacizumab):
• Den foretrukne kemoterapi blev givet i kombination med bevacizumab 10 mg / kg i.v. hver 2. uge (eller bevacizumab 15 mg / kg hver 3. uge i kombination med topotecan 1,25 mg / m2 på dag 1 "." 5 hver 3. uge).
Kvalificerede patienter havde epitelial æggestokkræft, æggelederkræft eller progressiv primær peritoneal kræft mindre end 6 måneder efter tidligere platinbaseret behandling bestående af mindst 4 behandlingsforløb.
Patienter skal have haft en forventet levetid på ≥12 uger og må ikke have haft tidligere strålebehandling mod bækken eller mave. De fleste af patienterne havde FIGO fase IIIC eller IV sygdom. Størstedelen af patienterne i begge arme havde en ECOG Performance Status (PS) på 0 (CT: 56,4% vs CT + BV: 61,2%). Procentdelen af patienter med ECOG PS på 1 eller ≥ 2 var 38,7% og 5,0% i CT -armen og 29,8% og 9,0% i CT + BV -armen. Raceinformation er tilgængelig for 29,3% af patienterne, og næsten alle patienter var kaukasiske. Gennemsnitsalderen for patienterne var 61,0 (interval: 25-84) år. I alt 16 patienter (4,4%) var i alderen> 75 år. De samlede afbrydelsesgrader på grund af bivirkninger var på 8,8% i CT-armen og 43,6% i CT + BV-armen (hovedsageligt på grund af bivirkninger af grad 2-3) og median tid til seponering i CT + BV-armen var 5,2 måneder mod 2,4 måneder i CT-armen. undergruppe af patienter> 65 år var 8,8% i CT -armen og 50,0% i CT + BV -armen. HR for PFS var henholdsvis 0,47 (95% CI: 0,35, 0,62) og 0,45 (95% CI: 0,31, 0,67) for aldersundergrupper.
Det primære endepunkt var progressionsfri overlevelse, mens de sekundære endepunkter omfattede objektiv responsrate og samlet overlevelse.Resultaterne er præsenteret i tabel 21.
Tabel 21 Effektresultater fra undersøgelse MO22224
Alle analyser præsenteret i denne tabel er lagdelte analyser.
* Datoafskæringsdatoen, hvormed den primære analyse blev udført, er 14. november 2011.
** Randomiserede patienter med målbar sygdom ved baseline.
*** Den endelige analyse af den samlede overlevelse blev udført, når 266 dødsfald blev observeret, svarende til 73,7% af de tilmeldte patienter.
Undersøgelsen nåede sit primære mål om at forbedre PFS. Sammenlignet med patienter behandlet med kemoterapi alene (paclitaxel, topotecan eller PLD) i forbindelse med platinresistent tilbagefald, patienter, der fik bevacizumab i en dosis på 10 mg / kg hver 2. uge (eller 15 mg / kg hver 3. uge, når de blev brugt i kombination med topotecan 1,25 mg / m2 på dag 1 "." 5 hver 3. uge) i kombination med kemoterapi og som fortsatte med at modtage bevacizumab indtil sygdomsprogression eller udvikling af uacceptabel toksicitet viste forbedring statistisk signifikant for PFS. Undersøgelsesanalyser af PFS og OS i kemoterapi -kohorten (paclitaxel, topotecan og PLD) er opsummeret i tabel 22.
Tabel 22: PFS og OS undersøgende analyse af kemoterapi kohorter.
Livmoderhalskræft
GOG-0240
Avastins effekt og sikkerhed i kombination med kemoterapi (paclitaxel og cisplatin eller paclitaxel og topotecan) ved behandling af patienter med vedvarende, tilbagevendende eller metastatisk livmoderhalskræft blev undersøgt i GOG-0240-forsøget, et fase III-studie. Randomiseret, fire- arm, open-label og multicenter.
I alt 452 patienter blev randomiseret til behandling med:
• Paclitaxel 135 mg / m2 i.v. over 24 timer på dag 1 og cisplatin 50 mg / m2 i.v. på dag 2, hver 3. uge (q3w); eller
Paclitaxel 175 mg / m2 i.v. over 3 timer på dag 1 og cisplatin 50 mg / m2 i.v. på dag 2 (q3w); eller
Paclitaxel 175 mg / m2 i.v. over 3 timer på dag 1 og cisplatin 50 mg / m2 i.v. på dag 1 (q3w)
• Paclitaxel 135 mg / m2 i.v. over 24 timer på dag 1 og cisplatin 50 mg / m2 i.v. på dag 2 + bevacizumab 15 mg / kg i.v. på dag 2 (q3w); eller
Paclitaxel 175 mg / m2 i.v. over 3 timer på dag 1 og cisplatin 50 mg / m2 i.v. på dag 2 + bevacizumab 15 mg / kg i.v. på dag 2 (q3w); eller
Paclitaxel 175 mg / m2 i.v. over 3 timer på dag 1 og cisplatin 50 mg / m2 i.v. på dag 1 + bevacizumab 15 mg / kg i.v. på dag 1 (q3w)
• Paclitaxel 175 mg / m2 i.v. over 3 timer på dag 1 og topotecan 0,75 mg / m2 i.v. over 30 minutter på dagene 1-3 (q3w)
• Paclitaxel 175 mg / m2 i.v. over 3 timer på dag 1 og topotecan 0,75 mg / m2 i.v. over 30 minutter på dagene 1-3 + bevacizumab 15 mg / kg i.v. på dag 1 (q3w).
Kvalificerede patienter havde vedvarende, tilbagevendende eller metastatisk pladecellecarcinom, adenosquamøst karcinom eller adenocarcinom i livmoderhalsen, der ikke var genstand for helbredende hensigt ved kirurgi og / eller strålebehandling og ikke var blevet forbehandlet med bevacizumab eller andre VEGF-hæmmere eller receptor-målrettende midler VEGF .
Medianalderen var 46,0 år (interval: 20-83) i gruppen kun kemoterapi og 48,0 år (interval: 22-85) i kemoterapien + Avastin-gruppen, med en alder over 65 år med henholdsvis 9,3% i gruppen behandlet alene med kemoterapi og 7,5% i gruppen behandlet med kemoterapi + Avastin.
De fleste af de 452 randomiserede patienter ved baseline var kaukasiske (80,0% i gruppen kun kemoterapi og 75,3% i kemoterapien + Avastin-gruppen), havde pladecellecarcinom (67,1% i kemoterapigruppen) og 69,6% i gruppen kemoterapi + Avastin-gruppe), persistens / tilbagefald af sygdom (83,6% i kemoterapigruppen og 82,8% i kemoterapien + Avastin-gruppen), 1-2 metastatiske steder (72,0% i kemoterapigruppen og 76,2% i gruppen kemoterapi + Avastin-gruppe), lymfeknudeinddragelse (50,2% i kemoterapigruppen og 56,4% i kemoterapien + Avastin-gruppen) og platinfrit interval ≥ 6 måneder (72,5% i kemoterapigruppen og 64,4% i gruppen kemoterapi + Avastin -gruppe).
Det primære effekt-endepunkt var samlet overlevelse. Sekundære effekt-endpoints omfattede progressionsfri overlevelse og objektiv responsrate. Resultater fra primær analyse og opfølgende analyse er vist som en funktion af Avastin-behandlingen. Og af den eksperimentelle behandling i tabel 23 og 24, henholdsvis.
Tabel 23 Effektresultater fra undersøgelse GOG-0240 med Avastin-baseret behandling
1 Estimater ifølge Kaplan-Meier
2 Patienter og procentdel af patienter med delvis respons (PR) eller komplet respons (CR) bekræftet som bedste samlede svar; procent beregnet på patienter med målbar sygdom ved baseline.
3 95% CI for en binomisk prøve i henhold til Pearson-Clopper-metoden
4IC ved 95% omtrentlige for forskellen mellem de to satser ifølge Hauck-Anderson-metoden
5 Log-rank test (stratificeret)
6L "primær analyse blev udført med en skæringsdato den 12. december 2012 og betragtes som en endelig analyse.
7 Opfølgningsanalysen blev udført med en skæringsdato den 7. marts 2014.
8P-værdi vises kun til beskrivende formål
Tabel 24 Samlede overlevelsesresultater fra undersøgelse GOG-0240 med undersøgelsesbehandling
1 Den primære analyse blev udført med en skæringsdato den 12. december 2012 og betragtes som den endelige analyse.
2 Opfølgningsanalyse blev udført med en skæringsdato den 7. marts 2014 Alle p-værdier er kun vist til beskrivende formål
Pædiatrisk population
Det Europæiske Lægemiddelagentur har frafaldet forpligtelsen til at indsende resultaterne af undersøgelser med bevacizumab i alle undergrupper af den pædiatriske population, i brystkræft, i tyktarm og rektal adenocarcinom, i lungekræft (mikrocytom og ikke-små celler), nyre- og nyrebekken karcinom (undtagen nefroblastom, nefroblastomatose, klar cellesarkom, meroblastisk nefrom, renalt medullært karcinom og rabdoid tumor i nyrerne), æggestokkræft (undtagen rabdomyosarkom og kimcelletumorer), æggelederkarcinom (eksklusive rabdomyosarcoma) (undtagen blastomer og sarkomer) og carcinom i livmoderhalsen og livmoderkroppen.
Ingen anticanceraktivitet blev observeret i to undersøgelser med i alt 30 børn i alderen> 3 år med tilbagefald eller progressivt gliom af høj kvalitet, når de blev behandlet med bevacizumab og irinotecan. Der er utilstrækkelig information til at bestemme sikkerhed og effekt af bevacizumab hos nyligt diagnosticerede børn med gliom af høj kvalitet.
I en enkeltarmsundersøgelse (PBTC-022), 18 børn med recidiverende eller progressive non-pontine gliom af høj kvalitet (herunder 8 med glioblastom [WHO grad IV], 9 med anaplastisk [grad III] astrocytom og 1 med anaplastisk oligodendrogliom [ Grad III]) blev behandlet med bevacizumab (10 mg / kg) med to ugers mellemrum og derefter med bevacizumab i kombination med CPT-11 (125-350 mg / m2) en gang hver anden uge indtil progression. Der var ingen objektive (delvise eller fuldstændige) radiologiske reaktioner (MacDonald -kriterier). Toksicitet og bivirkninger omfattede arteriel hypertension og træthed samt CNS -iskæmi med akut neurologisk underskud.
I en retrospektiv serie udført på en enkelt institution blev 12 børn med tilbagefaldende eller progressivt gliom af høj kvalitet (3 med WHO grad IV, 9 med grad III) behandlet i træk (2005 til 2008) med bevacizumab (10 mg / kg) og irinotecan (125 mg / m2) hver 2. uge. Der var 2 delvise svar og ingen komplette svar (MacDonald -kriterier).
05.2 Farmakokinetiske egenskaber
Farmakokinetiske data for bevacizumab indsamlet i ti kliniske undersøgelser udført hos patienter med solide maligniteter er tilgængelige. I alle kliniske undersøgelser blev bevacizumab administreret som en intravenøs infusion. Infusionshastigheden var afhængig af tolerabilitet med en indledende infusionsvarighed på 90 minutter. Den farmakokinetiske profil af bevacizumab var lineær ved doser fra 1 til 10 mg / kg.
Fordeling
Den typiske værdi for det centrale rumvolumen (Vc) var henholdsvis 2,73 l og 3,28 l for kvindelige og mandlige patienter, værdier i det område, der er beskrevet for IgG og andre monoklonale antistoffer. Typisk perifert rumvolumen (Vp) var 1,69 l og 2,35 l for henholdsvis kvindelige og mandlige patienter, når bevacizumab administreres med antineoplastiske midler. Efter korrektion for kropsvægt havde kønspatienter mandlige patienter en større Vc (+ 20%) end kvindelige patienter.
Biotransformation
Analyse af metabolismen af bevacizumab hos kaniner behandlet med en enkelt iv-dosis på 125I-bevacizumab afslørede en metabolisk profil svarende til den, der forventes for et naturligt IgG-molekyle, som ikke binder sig til VEGF. Metabolisme og eliminering af bevacizumab ligner den af endogent IgG, derfor primært gennem proteolytisk katabolisme i alle dele af kroppen, inklusive endotelceller og er ikke primært baseret på eliminering gennem nyrerne og leveren. Bindingen af IgG til FcRn -receptoren bestemmer beskyttelsen mod cellulær metabolisme og en lang terminal halvdel -liv.
Eliminering
Clearanceværdien var i gennemsnit 0,188 og 0,220 l / dag for henholdsvis kvindelige og mandlige patienter. Efter korrektion for kropsvægt havde mandlige patienter en højere clearance af bevacizumab (+ 17%) end kvindelige patienter. I forhold til bikompartimodellen er eliminationshalveringstiden 18 dage for en typisk kvindelig patient og 20 dage for en typisk mandlig patient.
Lave albuminværdier og en stor tumorbyrde er generelt indikatorer på sygdommens sværhedsgrad. Clearance af bevacizumab var cirka 30% hurtigere hos patienter med lavt serumalbumin og 7% hurtigere hos personer med stor tumorbyrde sammenlignet med en typisk patient med albuminværdier og gennemsnitlig tumorbyrde.
Farmakokinetik, især patientpopulationer
Befolkningens farmakokinetik blev analyseret for at evaluere virkningerne af demografiske karakteristika. Resultaterne af denne analyse afslørede ikke en signifikant forskel i bevacizumabs farmakokinetik baseret på alder.
Nyresvigt
Der er ikke udført undersøgelser for at undersøge bevacizumabs farmakokinetik hos patienter med nyreinsufficiens, da nyrerne ikke er et kritisk organ for metabolisme eller udskillelse af bevacizumab.
Leverinsufficiens
Der er ikke udført undersøgelser for at undersøge bevacizumabs farmakokinetik hos patienter med leverinsufficiens, da leveren ikke er et kritisk organ for metabolisme eller udskillelse af bevacizumab.
Pædiatrisk population
Farmacokinetikken af bevacizumab er undersøgt hos et begrænset antal pædiatriske patienter. De opnåede farmakokinetiske data tyder på, at distributionsvolumen og clearance af bevacizumab er sammenligneligt med dem hos voksne med solide tumorer.
05.3 Prækliniske sikkerhedsdata
I undersøgelser af op til 26 ugers varighed hos cynomolgus aber blev epifysisk dysplasi observeret hos unge dyr med åbne vækstplader ved gennemsnitlige serumkoncentrationer af bevacizumab under de gennemsnitlige terapeutiske serumkoncentrationer, der forventes hos mennesker. På kaniner har bevacizumab hæmmet sårhelingsprocessen ved doser under den foreslåede kliniske dosis, men virkningerne på sårhelingsprocessen var fuldstændig reversible.
Der er ikke udført undersøgelser for at evaluere bevacizumabs mutagene og kræftfremkaldende potentiale.
Der er ikke udført specifikke dyreforsøg for at evaluere effekten på fertilitet. Det er dog rimeligt at forvente en negativ indvirkning på kvindens fertilitet, da undersøgelser udført på dyr om toksicitet forbundet med administration af flere doser har vist en "hæmning af modning af ovariefollikler og en reduktion / fravær af corpora lutea, med den deraf følgende reduktion i vægten af æggestokkene og livmoderen, samt antallet af menstruationscyklusser.
Bevacizumab var embryotoksisk og teratogent hos kaninen. De observerede virkninger omfattede nedsat moder- og fostervægt, øget antal fosterresorptioner og øget forekomst af specifikke alvorlige misdannelser og fosterskelettet misdannelser. Dødelige følger, der påvirker fosteret, blev observeret ved alle testede doser; den administrerede laveste dosis resulterede i gennemsnitlige serumkoncentrationer cirka 3 gange højere end dem, der blev fundet hos mennesker efter administration af 5 mg / kg hver 2. uge. effekter.
06.0 LÆGEMIDDELOPLYSNINGER
06.1 Hjælpestoffer
Trehalose dihydrat
Natriumphosphat
Polysorbat 20
Vand til injektionsvæsker
06.2 Uforenelighed
Dette lægemiddel må ikke blandes med andre lægemidler end dem, der er nævnt i afsnit 6.6.
Når bevacizumab fortyndes med glucoseopløsninger (5%), observeres en koncentrationsafhængig nedbrydningsprofil.
06.3 Gyldighedsperiode
Hætteglas (lukket)
2 år.
Fortyndet medicin
Kemisk og fysisk stabilitet i brug er blevet påvist i 48 timer ved 2 ° C til 30 ° C i natriumchlorid 9 mg / ml (0,9%) injektionsvæske, opløsning. Mikrobiologisk, produktet skal bruges med det samme. Hvis brugen ikke er øjeblikkeligt, skal brugeren holdes ansvarlig for opbevaringstider og betingelser, som normalt ikke må overstige 24 timer ved en temperatur mellem 2 ° C og 8 ° C, medmindre fortynding er sket under kontrollerede og validerede aseptiske forhold.
06.4 Særlige opbevaringsforhold
Opbevares i køleskab (2 ° C-8 ° C).
Må ikke fryses.
Opbevar hætteglasset i den ydre karton for at beskytte det mod lys.
Opbevaringsbetingelser efter fortynding af lægemidlet, se pkt. 6.3.
06.5 Den umiddelbare emballages art og emballagens indhold
4 ml opløsning i et hætteglas (type I -glas) med en prop (butylgummi) indeholdende 100 mg bevacizumab.
16 ml opløsning i et hætteglas (type I -glas) med en prop (butylgummi) indeholdende 400 mg bevacizumab.
Pakke med 1 hætteglas.
06.6 Brugsanvisning og håndtering
Avastin skal fremstilles af en sundhedsperson med aseptisk teknik for at sikre steriliteten af den færdige løsning, der er fremstillet.
Den nødvendige mængde bevacizumab skal trækkes tilbage og fortyndes til det passende administrationsvolumen med natriumchlorid 9 mg / ml (0,9%) injektionsvæske, opløsning. Koncentrationen af den endelige bevacizumab -opløsning bør holdes inden for området 1,4 mg / ml til 16,5 mg / ml. I de fleste tilfælde kan den nødvendige mængde Avastin fortyndes med 0,9% natriumchloridopløsning til injektion til et samlet volumen på 100 ml.
Lægemidler bestemt til parenteral administration bør underkastes en visuel undersøgelse, inden de administreres for at udelukke tilstedeværelsen af partikler og tegn på farveændringer.
Der er ikke observeret uforligeligheder mellem Avastin og polyvinylchlorid eller polyolefin infusionsposer eller sæt.
Avastin er kun til engangsbrug, da produktet ikke indeholder konserveringsmidler. Ubrugt medicin og affald fra denne medicin skal bortskaffes i overensstemmelse med lokale regler.
07.0 INDEHAVER AF MARKEDSFØRINGSTILLADELSE
Roche registrering begrænset
6 Falcon Way
Shire Park
Welwyn Garden City
AL7 1TW
Storbritannien
08.0 MARKEDSFØRINGSTILLADELSESNUMMER
EU/1/04/300/001 - 100 mg/4 ml hætteglas
036680015
EU/1/04/300/002 - 400 mg/16 ml hætteglas
036680027
09.0 DATO FOR FØRSTE TILLADELSE ELLER FORNYELSE AF TILLADELSEN
Dato for første godkendelse: 12. januar 2005
Dato for sidste fornyelse: 14. januar 2015
10.0 DATO FOR REVISION AF TEKSTEN
Marts 2015







.jpg)