Aktive ingredienser: Fondaparinux (fondaparinuxnatrium)
Arixtra 1,5 mg / 0,3 ml injektionsvæske, opløsning
Arixtra indlægssedler er tilgængelige til pakningsstørrelser:- Arixtra 1,5 mg / 0,3 ml injektionsvæske, opløsning
- Arixtra 2,5 mg / 0,5 ml injektionsvæske, opløsning
- Arixtra 5 mg / 0,4 ml injektionsvæske, opløsning, Arixtra 7,5 mg / 0,6 ml injektionsvæske, opløsning, Arixtra 10 mg / 0,8 ml injektionsvæske, opløsning
Hvorfor bruges Arixtra? Hvad er det for?
Arixtra er et lægemiddel, der hjælper med at forhindre dannelse af blodpropper i blodkar (antitrombotisk middel).
Arixtra indeholder et stof kaldet fondaparinuxnatrium. Det virker ved at hæmme aktiviteten af koagulationsfaktor Xa ("ten-A") i blodet og derved forhindre dannelse af blodpropper (trombose) i blodkarrene.
Arixtra bruges til:
- forhindre dannelse af blodpropper i blodkarrene i benene eller lungerne efter ortopædisk kirurgi (såsom hofte- eller knæoperation) eller efter maveoperation
- forhindre dannelse af blodpropper under og umiddelbart efter en periode med begrænset mobilitet på grund af en akut sygdom
- behandling af blodpropper i benets overfladiske blodkar (overfladisk venetrombose).
Kontraindikationer Når Arixtra ikke bør bruges
Brug ikke Arixtra:
- hvis du er allergisk over for fondaparinuxnatrium eller et af de øvrige indholdsstoffer i denne medicin
- hvis du har kraftig blødning
- hvis du har en "bakteriel hjerteinfektion;
- hvis du har en meget alvorlig nyresygdom.
Fortæl det til din læge, hvis du mener, at noget af dette gælder for dig. I så fald bør du ikke bruge Arixtra.
Forholdsregler ved brug Det, du skal vide, før du tager Arixtra
Vær ekstra forsigtig med at bruge Arixtra:
Tal med din læge eller apotek, før du tager Arixtra:
- hvis du har risiko for ukontrolleret blødning (blødninger), som omfatter: mavesår blødningssygdom nylig hjerneblødning (intrakraniel blødning) nylig hjerne-, rygsøjle- eller øjenoperation
- hvis du har alvorlig leversygdom
- hvis du har en nyresygdom
- hvis du er 75 år eller ældre
- hvis du vejer mindre end 50 kg.
Fortæl det til din læge, hvis du mener, at noget af dette gælder for dig.
Børn og unge
Arixtra er ikke testet til brug hos børn og unge under 17 år.
Interaktioner Hvilke lægemidler eller fødevarer kan ændre virkningen af Arixtra
Fortæl det til din læge eller apotek, hvis du tager anden medicin eller har brugt det for nylig. Dette inkluderer også dem, der er købt uden recept. Nogle andre lægemidler kan påvirke måden Arixtra virker på eller kan blive påvirket af Arixtra.
Advarsler Det er vigtigt at vide, at:
Graviditet og amning
Arixtra bør ikke ordineres til gravide, medmindre det er specifikt nødvendigt. Amning anbefales ikke, mens du tager Arixtra. Hvis du er gravid eller ammer, tror at du er gravid eller planlægger at blive gravid, skal du spørge din læge eller apotek til råds, før du bruger denne medicin.
Arixtra indeholder natrium
Hver dosis af dette lægemiddel indeholder mindre end 23 mg natrium og er derfor i det væsentlige natriumfri.
Arixtra sprøjten indeholder latex
Sprøjtenålens dæksel indeholder latex, der kan forårsage allergiske reaktioner hos latexfølsomme personer.
- Fortæl det til din læge, hvis du har en latexallergi, før du behandles med Arixtra.
Dosis, metode og administrationstidspunkt Sådan bruges Arixtra: Dosering
Brug altid dette lægemiddel nøjagtigt som din læge eller apotek har fortalt dig. Hvis du er i tvivl, skal du kontakte din læge eller apotek.
Den anbefalede dosis er 2,5 mg en gang dagligt, der skal injiceres på omtrent samme tidspunkt hver dag.
Hvis du har nyresygdom, kan dosis reduceres til 1,5 mg en gang dagligt.
Sådan gives Arixtra
- Arixtra gives ved injektion under huden (subkutant) i en hudfold i det nedre del af maven. Sprøjterne er fyldt med den nøjagtige dosis, der er nødvendig. Doseringssprøjterne på 2,5 mg og 1,5 mg er forskellige. For en "brugsanvisning" punkt for punkt, se slutningen af arket.
- Injicer ikke Arixtra i musklen.
Hvor lang tid skal Arixtra tages
Du skal fortsætte Arixtra -behandlingen, så længe din læge har ordineret det, da Arixtra forhindrer udviklingen af alvorlige sygdomme
Overdosering Hvad skal jeg gøre, hvis du har taget for meget Arixtra
Hvis du injicerer for meget Arixtra
Kontakt din læge eller apotek så hurtigt som muligt for at få råd, da dette øger risikoen for blødning.
Hvis du har glemt at tage Arixtra
- Giv dosis, så snart du husker det. Injektér ikke en dobbelt dosis for at kompensere for en glemt dosis.
- Hvis du ikke er sikker på, hvad du skal gøre, skal du kontakte din læge eller apotek.
Stop ikke med at bruge Arixtra uden lægehjælp
Hvis du stopper behandlingen tidligere, end din læge har ordineret, risikerer du at udvikle en blodprop i en vene i dine ben eller i lungerne. Kontakt din læge eller apotek, inden behandlingen stoppes.
Spørg din læge eller apotek, hvis du har yderligere spørgsmål om brugen af dette lægemiddel.
Bivirkninger Hvad er bivirkningerne af Arixtra
Ligesom al anden medicin kan denne medicin forårsage bivirkninger, men ikke alle får bivirkninger.
Betingelser, som det er nødvendigt at bede om hjælp til
Alvorlige allergiske reaktioner (anafylaksi): er meget sjældne hos mennesker, der tager Arixtra (op til 1 ud af 10.000 mennesker). Symptomer omfatter:
- hævelse, nogle gange i ansigt eller mund (angioødem), hvilket forårsager synke- eller vejrtrækningsbesvær
- falde sammen.
Kontakt straks din læge, hvis du oplever sådanne symptomer. Stop med at tage Arixtra.
Almindelige bivirkninger
De kan påvirke mere end en ud af 100 mennesker, der behandles med Arixtra:
- blødning (f.eks. på operationsstedet, fra et allerede eksisterende mavesår, fra næsen, fra tandkødet)
- anæmi (en reduktion i antallet af røde blodlegemer).
Ikke almindelige bivirkninger
De kan påvirke op til en ud af 100 mennesker, der behandles med Arixtra:
- blå mærker eller hævelse (ødem)
- kvalme eller opkastning
- brystsmerter
- stakåndet
- rødme eller kløe
- væske, der siver fra såret efter operationen
- feber
- fald eller stigning i antallet af blodplader (blodlegemer nødvendige for koagulation)
- stigning i nogle stoffer (enzymer) produceret af leveren
Sjældne bivirkninger
De kan ramme op til 1 ud af 1.000 personer, der behandles med Arixtra:
- allergiske reaktioner (herunder kløe, hævelse, udslæt)
- indre hjerne eller abdominal blødning
- angst eller forvirring
- hovedpine
- besvimelse eller svimmelhed, lavt blodtryk
- døsighed eller træthed
- hedeture
- hoste
- smerter i benene eller maven
- diarré eller forstoppelse
- dårlig fordøjelse
- sårinfektion
- stigning i bilirubin (et stof produceret af leveren) i blodet
- reduktion af kalium i blodet.
Indberetning af bivirkninger
Tal med din læge eller apotek, hvis du får bivirkninger, herunder mulige bivirkninger, som ikke er nævnt i denne indlægsseddel. Du kan også indberette bivirkninger direkte via det nationale rapporteringssystem anført i tillæg V. Ved at rapportere bivirkninger kan du hjælpe med at give mere information om sikkerheden ved dette lægemiddel.
Udløb og opbevaring
- Opbevar denne medicin utilgængeligt for børn.
- Opbevares under 25 ° C. Må ikke fryses.
- Arixtra må ikke opbevares i køleskab.
Brug ikke denne medicin:
- efter den udløbsdato, der er angivet på etiketten og æsken
- hvis du bemærker tilstedeværelsen af partikler i opløsningen, eller hvis opløsningen har en unormal farve
- hvis du bemærker, at sprøjten er beskadiget
- hvis du har åbnet en sprøjte og ikke skal bruge den med det samme.
Bortskaffelse af sprøjter:
Smid ikke medicin eller sprøjte i spildevand eller husholdningsaffald. Spørg din apotek om, hvordan du skal smide medicin, du ikke længere bruger. Dette vil hjælpe med at beskytte miljøet.
Pakningens indhold og andre oplysninger
Hvad Arixtra indeholder
- Det aktive stof er 1,5 mg fondaparinuxnatrium i 0,3 ml injektionsvæske, opløsning.
- Øvrige indholdsstoffer er natriumchlorid, vand til injektionsvæsker og saltsyre og / eller natriumhydroxid til pH -justering.
Arixtra indeholder ikke produkter af animalsk oprindelse.
Beskrivelse af hvordan Arixtra ser ud og pakningens indhold
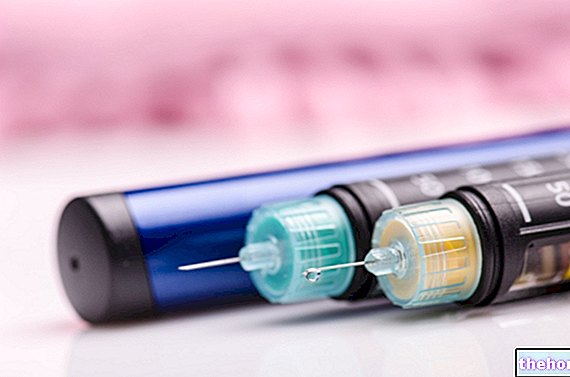
Arixtra er en klar og farveløs injektionsvæske, opløsning. Den leveres med en fyldt, engangssprøjte, komplet med et beskyttelsessystem, der er designet til at beskytte mod utilsigtede nålepinde efter brug. Den fås i pakninger med 2, 7, 10 og 20 fyldte sprøjter (ikke alle pakningsstørrelser kan blive markedsført).
PUNKT FOR PUNKT VED AT BRUGE ARIXTRA GUIDE
Brugsanvisning
Disse instruktioner er gyldige for begge typer sprøjter (automatisk og manuelt nålebeskyttelsessystem)
Hvor instruktionerne for hver sprøjte er forskellige, er dette tydeligt angivet.
1. Vask dine hænder grundigt med sæbe og vand, og tør dem derefter med et håndklæde.
2. Tag sprøjten ud af kassen, og kontroller, at:
- udløbsdatoen ikke er gået
- opløsningen er klar og farveløs og indeholder ikke partikler
- sprøjten ikke er blevet åbnet eller beskadiget
3. Sid eller læg dig ned i en behagelig stilling.
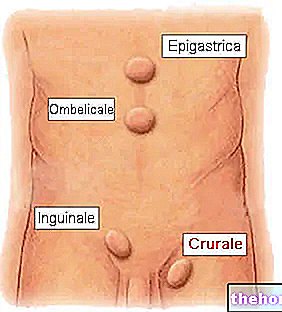
Vælg et punkt i det nedre maveområde, mindst 5 cm under navlen
Skift venstre og højre side af det nedre del af maven med hver injektion. Dette hjælper med at reducere ubehag på injektionsstedet.
Hvis det ikke er muligt at injicere det nedre del af maven, skal du kontakte din sygeplejerske eller læge for at få råd.
4. Rengør injektionsstedet med en spritserviet.
5. Fjern kanyledækslet ved først at vride det og derefter trække det lige væk fra sprøjtens krop. Fjern hætten.
Vigtig note
- Rør ikke ved nålen, og sørg for, at den ikke kommer i kontakt med andre overflader, før du injicerer.
- Tilstedeværelsen af en lille luftboble i sprøjten er normal. Forsøg ikke at fjerne små luftbobler før injektion for at sikre, at du ikke mister noget produkt.
6. Klem let det desinficerede område af huden for at danne en fold. Hold folden mellem tommelfinger og pegefinger under hele injektionen
7. Hold sprøjten fast mellem fingrene.
Sæt vinkelret (i en 90 ° vinkel) hele nålens længde ind i hudfolden
8. Injicér ALT indholdet i sprøjten ved at skubbe stemplet så langt ned som muligt
Sprøjte med automatisk system
9. Slip stemplet, og nålen vil automatisk trække sig tilbage fra huden ind i sikkerhedsbøsningen, hvor den forbliver permanent lukket
Sprøjte med manuelt system
9. Efter injektion skal du holde sprøjten i den ene hånd, mens du holder i sikkerhedsbøsningen, bruge den anden hånd til at holde i håndtaget og trække fast tilbage. Dette låser ærmen op. Skub ærmen gennem sprøjtens krop, indtil den klikker på plads over nålen
Bortskaf ikke den brugte sprøjte sammen med husholdningsaffald. Smid den brugte sprøjte efter instruktionerne fra din læge eller apotek
Indlægsseddel: AIFA (Italian Medicines Agency). Indhold offentliggjort i januar 2016. De foreliggende oplysninger er muligvis ikke opdaterede.
For at få adgang til den mest opdaterede version er det tilrådeligt at få adgang til webstedet AIFA (Italian Medicines Agency). Ansvarsfraskrivelse og nyttige oplysninger.
01.0 LÆGEMIDLETS NAVN
ARIXTRA 1,5 MG / 0,3 ML
02.0 KVALITATIV OG KVANTITATIV SAMMENSÆTNING
Hver fyldt injektionssprøjte (0,3 ml) indeholder 1,5 mg fondaparinuxnatrium.
Hjælpestoffer med kendte virkninger: Indeholder mindre end 1 mmol natrium (23 mg) pr. Dosis og er derfor i det væsentlige natriumfrit.
Den fulde liste over hjælpestoffer findes i afsnit 6.1.
03.0 LÆGEMIDDELFORM
Injicerbar løsning.
Opløsningen er en klar, farveløs væske.
04.0 KLINISKE OPLYSNINGER
04.1 Terapeutiske indikationer
Forebyggelse af venøse tromboemboliske episoder (VTE) hos voksne, der gennemgår større ortopædkirurgi i underekstremiteterne, såsom hoftebrud, større knæoperationer eller hofteudskiftningskirurgi.
Forebyggelse af venøse tromboemboliske episoder (VTE) hos voksne, der gennemgår en abdominal kirurgi, anses for at have stor risiko for tromboemboliske komplikationer, f.eks. Patienter, der gennemgår en abdominal operation for kræft (se pkt.5.1).
Forebyggelse af venøse tromboemboliske episoder (VTE) hos medicinsk relevante voksne, der anses for at have høj risiko for VTE, og som er immobiliseret på grund af en akut tilstand som hjertesvigt og / eller akut luftvejssygdom og / eller akut inflammatorisk sygdom eller infektion.
Behandling af voksne med akut spontan symptomatisk overfladisk venetrombose i underekstremiteterne i mangel af samtidig dyb venetrombose (se pkt. 4.2 og 5.1).
04.2 Dosering og indgivelsesmåde
Dosering
Patienter, der gennemgår større ortopædisk eller abdominal kirurgi
Den anbefalede dosis fondaparinux er 2,5 mg administreret en gang dagligt efter operationen ved subkutan injektion.
Startdosis bør administreres 6 timer efter operationens afslutning, når hæmostase er sikret.
Behandlingen bør fortsættes, indtil risikoen for venøs tromboemboli falder, normalt indtil patienten genoptager gåingen, mindst 5-9 dage efter operationen.Erfaring viser, at risikoen for VTE ved patienter, der gennemgår hoftebrud, fortsætter ud over 9 dage efter operationen. Hos disse patienter bør brug af forlænget fondaparinux -profylakse overvejes i op til yderligere 24 dage (se pkt.5.1).
Medicinsk relevante patienter, der har stor risiko for tromboemboliske komplikationer baseret på en individuel risikovurdering
Den anbefalede dosis fondaparinux er 2,5 mg én gang dagligt administreret ved subkutan injektion. Behandling, der varer 6-14 dage, er blevet klinisk undersøgt hos medicinsk relevante patienter (se pkt.5.1).
Behandling af overfladisk venetrombose
Den anbefalede fondaparinux -dosis er 2,5 mg pr. Dag, administreret ved subkutan injektion. Patienter, der er berettiget til behandling med fondaparinux 2,5 mg, skal udvise spontan, akut, symptomatisk og isoleret overfladisk venøs trombose i underekstremiteterne, mindst 5 cm i længden og dokumenteret ved ultralyd eller andre fysiske undersøgelser. Behandlingen bør påbegyndes hurtigst muligt umiddelbart efter diagnosen og efter udelukkelse af samtidig dyb venetrombose (DVT) eller overfladisk venetrombose inden for 3 cm fra sapheno-femoral kryds. Behandlingen bør fortsættes i mindst 30 dage og op til maksimalt 45 dage hos patienter med høj risiko for tromboemboliske komplikationer (se pkt. 4.4 og 5.1).
Patienter bør anbefales at injicere produktet selv, når de efter lægens vurdering er villige og i stand til at gøre det. Læger bør give klare instruktioner til selvinjektion.
• Patienter, der skal opereres eller andre invasive procedurer
Hos patienter med overfladisk venetrombose, der skal opereres eller andre invasive procedurer, bør fondaparinux, hvor det er muligt, ikke administreres i løbet af 24 timer før operationen. Fondaparinux -behandling kan genstarte mindst 6 timer efter operationen. Kirurgisk, forudsat at hæmostase har været opnået.
Særlige kategorier af patienter
Hos patienter, der opereres, kræver administrationstiden for den første fondaparinux -injektion streng overholdelse hos patienter ≥ 75 år og / eller nyreinsufficiens med en kreatininclearance mellem 20 og 50 ml / min.
Den første administration af fondaparinux bør gives tidligst 6 timer efter operationens afslutning Injektionen bør ikke gives uden hæmostase fastslået (se pkt. 4.4).
Nyresvigt -
• VTE -forebyggelse - Fondaparinux bør ikke anvendes til patienter med kreatininclearance 50 ml / min).
• Behandling af overfladisk venetrombose - Fondaparinux bør ikke anvendes til patienter med kreatininclearance 50 ml / min). Sikkerheden og effekten af 1,5 mg er ikke undersøgt (se pkt. 4.4).
Leversvigt -
• VTE -forebyggelse - Ingen dosisjustering er nødvendig hos patienter med let eller moderat nedsat leverfunktion. Hos patienter med svært nedsat leverfunktion bør fondaparinux anvendes med forsigtighed, da det ikke er undersøgt i denne patientgruppe (se pkt. 4.4 og 5.2).
• Behandling af overfladisk venetrombose - Sikkerhed og effekt af fondaparinux er ikke undersøgt hos patienter med svært nedsat leverfunktion, derfor anbefales det ikke at bruge fondaparinux til disse patienter (se pkt. 4.4).
Pædiatrisk population - Fondaparinux anbefales ikke til børn under 17 år på grund af mangel på data om sikkerhed og effekt.
Lav kropsvægt
• VTE -forebyggelse - Patienter med blødende kropsvægt. Elimineringen af fondaparinux falder med vægten. Fondaparinux bør anvendes med forsigtighed til disse patienter (se pkt. 4.4).
• Behandling af overfladisk venetrombose - Sikkerhed og effekt af fondaparinux er ikke undersøgt hos patienter, der vejer mindre end 50 kg, og derfor anbefales fondaparinux ikke til brug hos disse patienter (se pkt. 4.4).
Indgivelsesmåde
Fondaparinux skal administreres ved dyb subkutan injektion, hvor patienten ligger i ryggen. Injektionsstedet skal skifte mellem venstre og højre anterolaterale side og mellem venstre og højre posterolaterale side af bugvæggen. For at undgå tab af medicin ved brug af den fyldte injektionssprøjte må luftbobler ikke udledes fra sprøjten før injektion. Hele nålens længde skal indsættes vinkelret i en hudfold holdt mellem tommelfinger og pegefinger; hudfolden skal opretholdes i løbet af injektionen.
For yderligere instruktioner om brug og bortskaffelse, se afsnit 6.6.
04.3 Kontraindikationer
- Kendt overfølsomhed over for det aktive stof eller over for et eller flere af hjælpestofferne anført i pkt.6.1
- igangværende blødning, klinisk signifikant
- akut bakteriel endokarditis
- alvorlig nyreinsufficiens defineret som kreatininclearance
04.4 Særlige advarsler og passende forholdsregler ved brug
Fondaparinux er kun beregnet til subkutan brug. Må ikke administreres intramuskulært.
Blødninger
Fondaparinux bør anvendes med forsigtighed til patienter, der har en øget risiko for blødning, f.eks. Dem med medfødte eller erhvervede blødningsforstyrrelser (f.eks. Trombocyttal 3), aktiv ulcerøs mave -tarmsygdom og nylig eller kort tid efter hjerne-, spinal- eller intrakraniel blødning. Oftalmisk og i særlige patientgrupper som angivet nedenfor.
• Til forebyggelse af VTE - Midler, der kan øge risikoen for blødning, bør ikke administreres samtidigt med fondaparinux. Sådanne stoffer omfatter desirudin, fibrinolytiske midler, GP IIb / IIIa -receptorantagonister, heparin, heparinoider eller lavmolekylær heparin (LMWH). Hvis det er påkrævet, bør samtidig behandling med vitamin K -antagonister administreres i henhold til anvisningerne i afsnit 4.5. Andre trombocytplader (acetylsalicylsyre, dipyridamol, sulfinpyrazon, ticlopidin eller clopidogrel) og NSAID'er bør anvendes med forsigtighed. Hvis samadministration er afgørende, er der behov for tæt overvågning.
• Til behandling af overfladisk venetrombose - Fondaparinux bør bruges med forsigtighed til patienter, der får samtidig medicin, der øger risikoen for blødning.
Patienter med overfladisk venetrombose
Tilstedeværelsen af overfladisk venøs trombose i en afstand større end 3 cm fra sapheno-femoral krydset skal bekræftes, før behandling påbegyndes med fondaparinux, og tilstedeværelsen af DVT skal udelukkes ved kompressionsultralyd (CUS) eller andre objektive metoder. Der er ingen data om anvendelse af fondaparinux 2,5 mg til patienter med overfladisk venetrombose forbundet med samtidig DVT eller med overfladisk venetrombose inden for 3 cm fra sapheno-femoral junction (se pkt. 4.2 og 5.1).
Sikkerheden og effekten af fondaparinux 2,5 mg er ikke undersøgt i følgende grupper: patienter med overfladisk venetrombose efter sklerosebehandling eller som en konsekvens af en intravenøs linje, patienter med en historie med overfladisk venetrombose inden for de foregående 3 måneder, patienter med en historie med venøs tromboembolisk sygdom inden for de foregående 6 måneder eller patienter med en aktiv tumor (se pkt. 4.2 og 5.1).
Spinal / epidural anæstesi
Hos patienter, der gennemgår større ortopædkirurgi, ved samtidig brug af fondaparinux og spinal / epidural anæstesi eller spinal punktering, kan forekomsten af epidural eller spinal hæmatom, der kan resultere i langvarig eller permanent lammelse, ikke udelukkes.Risikoen for disse sjældne hændelser kan øges med den postoperative brug af indbyggede epiduralkateter eller med samtidig brug af andre lægemidler, der virker på hæmostase.
Ældre patienter
Den ældre befolkning har en øget risiko for blødning. Da nyrefunktionen generelt falder med alderen, kan ældre patienter vise reduceret eliminering og øget eksponering for fondaparinux (se pkt. 5.2) Fondaparinux bør anvendes med forsigtighed til ældre patienter (se pkt.4.2).
Lav kropsvægt
• VTE -forebyggelse - Patienter med kropsvægt
• Behandling af overfladisk venetrombose - Der er ingen kliniske data til rådighed for brug af fondaparinux til behandling af overfladisk venetrombose hos patienter, der vejer mindre end 50 kg.Fondaparinux anbefales derfor ikke til behandling af overfladisk venetrombose hos disse patienter (se pkt. 4.2).
Nyresvigt
• VTE -forebyggelse - Fondaparinux er kendt for hovedsageligt at blive udskilt af nyrerne. Patienter med kreatininclearance
• Behandling af overfladisk venetrombose - Fondaparinux må ikke anvendes til patienter med kreatininclearance
Alvorlig leverinsufficiens
• VTE -forebyggelse - Ingen fondaparinux dosisjustering er nødvendig. Anvendelse af fondaparinux til patienter med alvorlig leverinsufficiens bør dog overvejes med forsigtighed på grund af en øget risiko for blødning på grund af mangel på koagulationsfaktorer hos patienter med alvorlig leverinsufficiens (se pkt.4.2).
• Behandling af overfladisk venetrombose - Der er ingen kliniske data tilgængelige for brug af fondaparinux til behandling af overfladisk venetrombose hos patienter med
alvorlig leverinsufficiens. Derfor anbefales fondaparinux ikke til behandling af overfladisk venetrombose hos disse patienter (se pkt.4.2).
Patienter med heparininduceret trombocytopeni
Fondaparinux bør anvendes med forsigtighed til patienter med en historie med heparininduceret trombocytopeni (HIT). Effekten og sikkerheden af fondaparinux hos patienter med type II HIT er ikke formelt undersøgt Fondaparinux binder ikke til koagulationsfaktor 4 og krydsreagerer ikke med plasmaet hos patienter med type II HIT Sjældne spontane rapporter om HIT er modtaget hos patienter behandlet med fondaparinux Til dato er der ikke fastslået en årsagssammenhæng mellem fondaparinux -behandling og starten af HIT.
Latex allergi
Nåledækslet på den fyldte sprøjte indeholder tør naturgummilatex, der kan forårsage allergiske reaktioner hos latexfølsomme personer.
04.5 Interaktioner med andre lægemidler og andre former for interaktion
Samtidig administration af fondaparinux og stoffer, der kan øge risikoen for blødning, øger risikoen for blødning (se pkt. 4.4).
Orale antikoagulantia (warfarin), trombocythæmmere (acetylsalicylsyre), NSAID (piroxicam) og digoxin interagerede ikke med fondaparinux farmakokinetik. Dosen af fondaparinux (10 mg) i interaktionsundersøgelserne var højere end den anbefalede dosis for de aktuelle indikationer. Fondaparinux påvirker hverken warfarins INR -aktivitet eller blødningstiden under behandling med acetylsalicylsyre eller piroxicam, eller digoxins steady state -farmakokinetik.
Fortsættelse af behandlingen med et andet antikoagulerende lægemiddel
Hvis fortsat behandling skal startes med heparin eller LMWH, bør den første injektion som hovedregel gives 1 dag efter den sidste fondaparinux -injektion.
Hvis det er nødvendigt at fortsætte behandlingen med en vitamin K -antagonist, bør fondaparinux -behandlingen fortsættes, indtil den etablerede INR -værdi er nået.
04.6 Graviditet og amning
Graviditet
Der er utilstrækkelige data om anvendelse af fondaparinux under graviditet. Dyrestudier er utilstrækkelige med hensyn til virkninger på graviditet, embryonisk / fosterudvikling, fødsel og postnatal udvikling på grund af begrænset eksponering. Fondaparinux bør ikke ordineres til gravide, medmindre det er strengt nødvendigt.
Amning
Fondaparinux udskilles i rottemælk, men det vides ikke, om fondaparinux udskilles i modermælk. Amning anbefales ikke under fondaparinux -behandling. Oral absorption af spædbarnet er dog usandsynlig.
Fertilitet
Der er ingen tilgængelige data om effekten af fondaparinux på menneskelig fertilitet Dyrestudier har ikke vist nogen effekt på fertiliteten.
04.7 Virkninger på evnen til at føre motorkøretøj og betjene maskiner
Der er ikke udført undersøgelser af evnen til at føre motorkøretøj eller betjene maskiner.
04.8 Bivirkninger
De hyppigst rapporterede alvorlige bivirkninger ved fondaparinux er blødningskomplikationer (på forskellige steder, herunder sjældne tilfælde af intrakraniel / intracerebral og retroperitoneal blødning) og anæmi. Fondaparinux bør anvendes med forsigtighed til patienter, der har en øget risiko for blødning (se pkt. 4.4).
Sikkerheden ved fondaparinux 2,5 mg blev evalueret hos 3.595 patienter, der gennemgik en større ortopædkirurgi i de nedre lemmer behandlet i op til 9 dage, hos 327 patienter, der blev opereret i hoftebrud i 3 uger efter en indledende profylakse på 1 uge, hos 1.407 patienter, der blev underkastet abdominal kirurgi behandlet i op til 9 dage og hos 425 medicinsk relevante patienter (ikke under kirurgisk behandling) med risiko for tromboemboliske komplikationer behandlet i op til 14 dage.
Bivirkninger rapporteret af forskere som i det mindste muligvis relateret til fondaparinux er præsenteret inden for hver frekvensgruppe (meget almindelig ≥1 / 10; almindelig: ≥ 1/100,
I andre undersøgelser eller efter markedsføring er der rapporteret om sjældne tilfælde af intrakraniel / intracerebral og retroperitoneal blødning.
Indberetning af formodede bivirkninger
Rapportering af formodede bivirkninger, der opstår efter godkendelse af lægemidlet, er vigtig. Det giver mulighed for løbende overvågning af fordelene / risikobalancen for lægemidlet. Sundhedspersonale anmodes om at rapportere alle formodede bivirkninger via det nationale rapporteringssystem. "Bilag V.
04.9 Overdosering
Doser af fondaparinux ud over det anbefalede regime kan føre til en øget risiko for blødning. Der er ingen kendte modgift mod fondaparinux.
Overdosering forbundet med blødningskomplikationer skal indebære afbrydelse af behandlingen og søgning af den primære årsag. Passende behandling såsom kirurgisk hæmostase, blodtransfusion, frisk plasmatransfusion, plasmaferese bør overvejes.
05.0 FARMAKOLOGISKE EGENSKABER
05.1 Farmakodynamiske egenskaber
Farmakoterapeutisk klassifikation: antitrombotiske midler.
ATC -kode: B01AX05.
Farmakodynamiske virkninger
Fondaparinux er en syntetisk og selektiv hæmmer af aktiveret faktor X (Xa). Den antitrombotiske aktivitet af fondaparinux er resultatet af selektiv inhibering af faktor Xa medieret af antithrombin III (ATIII). Ved selektiv binding til ATIII forbedrer fondaparinux (cirka 300 gange) den naturlige neutralisering af faktor Xa med ATIII. Af faktor Xa afbryder blodet koagulationskaskade og hæmmer både trombindannelse og trombudvikling Fondaparinux inaktiverer ikke thrombin (aktiveret faktor II) og har ingen effekt på blodplader.
Ved 2,5 mg dosis påvirker fondaparinux ikke rutinemæssige koagulationstests såsom aktiveret partiel tromboplastintid (aPTT), aktiveret koagulationstid (ACT) eller protrombintid (PT) / International Normalized Ratio (INR) i plasma, heller ikke blødningstid eller fibrinolytisk Der er dog kun modtaget sjældne rapporter om forlængelse af aPTT.
Fondaparinux krydsreagerer ikke med serum fra patienter med heparininduceret trombocytopeni.
Kliniske undersøgelser
Forebyggelse af venøs tromboemboli (VTE) hos patienter, der gennemgår en større ortopædkirurgi i de nedre lemmer, der er behandlet i op til 9 dage: fondaparinux kliniske plan var designet til at demonstrere fondaparinux effektivitet i forebyggelsen af venøse tromboemboliske hændelser (VTE), dvs. trombose proksimal og distal deep vene (DVT) og lungeemboli (PE) hos patienter, der gennemgår større ortopædkirurgi i underekstremiteterne, såsom hoftebrud, større knæoperationer eller hofteudskiftningskirurgi. I fase II og III kontrollerede kliniske forsøg blev over 8.000 patienter undersøgt ( hoftebrud - 1.711, hofteudskiftning - 5829, større knæoperationer - 1.367). Fondaparinux 2,5 mg en gang dagligt startede 6-8 timer efter operationen blev sammenlignet med enoxaparin 40 mg en gang dagligt startet 12 timer før operationen eller 30 mg to gange dagligt orno startede 12-24 timer efter operationen.
I en samlet analyse af disse undersøgelser var den anbefalede dosisregime af fondaparinux versus enoxaparin forbundet med et signifikant fald (54% -95% CI, 44%; 63%) i forekomsten af VTE estimeret op til 11. dag efter operationen, uanset af den type operation, der udføres. Størstedelen af "endepunkt" -hændelser blev diagnosticeret med en forudbestemt venografi og var hovedsageligt sammensat af distal DVT, men forekomsten af proksimal DVT var også signifikant reduceret. Forekomsten af symptomatisk VTE, inklusive PE, var ikke signifikant forskellig mellem behandling. grupper.
I undersøgelserne versus enoxaparin 40 mg én gang dagligt startet 12 timer før operationen blev der observeret alvorlig blødning hos 2,8% af patienterne behandlet med fondaparinux i den anbefalede dosis sammenlignet med 2,6% med enoxaparin.
Forebyggelse af venøs tromboemboli (VTE) hos patienter, der gennemgår hoftebrudskirurgi behandlet i op til 24 dage efter den første 1-ugers profylakse: I et dobbeltblindet, randomiseret klinisk forsøg blev 737 patienter behandlet med fondaparinux 2,5 mg én gang dagligt i 7 ± 1 dage efter hoftebrudskirurgi. Ved slutningen af denne periode blev 656 patienter randomiseret til at modtage fondaparinux 2,5 mg én gang dagligt eller placebo i yderligere 21 ± 2 dage. Fondaparinux gav en signifikant reduktion i den samlede forekomst af VTE sammenlignet med placebo [3 patienter (1,4%) henholdsvis 77 patienter (35%)]. Størstedelen (70/80) af de rapporterede VTE -episoder var tilfælde af asymptomatisk DVT påvist flebografisk Fondaparinux gav også en signifikant reduktion i forekomsten af symptomatisk VTE (henholdsvis DVT og / eller PE) [1 (0,3%) versus 9 (2,7%) patienter inklusive 2 dødelige PE rapporteret i placebogruppen. Alvorlig blødning, alle kirurgiske og ingen dødelig, blev observeret hos 8 patienter (2,4%) behandlet med fondaparinux 2,5 mg sammenlignet med 2 (0,6%) med placebo.
Forebyggelse af venøse tromboemboliske episoder (VTE) hos patienter, der gennemgår abdominal kirurgi, der anses for at have stor risiko for tromboemboliske komplikationer, såsom patienter, der gennemgår en abdominal kirurgi for kræft: I et dobbeltblind klinisk studie blev 2.927 patienter randomiseret til at modtage fondaparinux 2, 5 mg en gang dagligt eller dalteparin 5000 IE en gang dagligt, ved en præoperativ injektion på 2500 IE og en første postoperativ injektion på 2500 IE i 7 + 2 dage. De vigtigste operationssteder var kolorektal, gastrisk, hepatisk, cholecystektomi eller andre galdeinterventioner. 99 procent af patienterne blev opereret efter kræft. Patienterne blev gennemført urologisk (undtagen nyre) eller gynækologisk kirurgi., Laparoskopisk eller vaskulær kirurgi blev ikke inkluderet i undersøgelsen .
I denne undersøgelse var forekomsten af total VTE 4,6% (47 / 1,027) med fondaparinux sammenlignet med 6,1% (62 / 1,021) med dalteparin: reduktion i ulige forhold (95% CI) = - 25,8% (-49,7%, 9,5%). Forskellen i hyppigheden af total VTE mellem behandlingsgrupper, som ikke var statistisk signifikant, skyldtes hovedsageligt reduktionen i distal DVT. Forekomsten af symptomatisk DVT var ens mellem de to behandlingsgrupper: 6 patienter (0,4% ) i fondaparinux -gruppen versus 5 patienter (0,3%) i dalteparin -gruppen. I den store undergruppe af patienter, der gennemgik kræftoperation (69% af patientpopulationen), var frekvensen af VTE 4,7% i fondaparinux -gruppen mod 7,7% i dalteparingruppen.
Alvorlig blødning blev observeret hos 3,4% af fondaparinux-behandlede patienter og 2,4% af den dalteparin-behandlede gruppe.
Forebyggelse af venøse tromboemboliske episoder (VTE) hos medicinsk relevante patienter med høj risiko for tromboemboliske komplikationer på grund af nedsat mobilitet under akut sygdom: I et randomiseret dobbeltblindet klinisk forsøg blev 839 patienter behandlet i 6 til 14 dage med fondaparinux 2,5 mg én gang dagligt eller med placebo. Denne undersøgelse omfattede medicinsk relevante akutte patienter i alderen ≥ 60 år, som forventedes at være sengeliggende i mindst fire dage og indlagt på hospital for NYHA klasse III / IV hjertesvigt og / eller akut luftvejssygdom. Og / eller akut infektiøs eller inflammatorisk patologi. Fondaparinux sammenlignet med placebo reducerede signifikant den samlede forekomst af VTE [henholdsvis 18 patienter (5,6%) versus 34 patienter (10,5%)]. Størstedelen af hændelserne var asymptomatisk distal DVT. Fondaparinux reducerede også signifikant forekomsten af PE, der blev betragtet som dødelig [0 patienter (henholdsvis 0,0%) vs 5 patienter (1,2%)]. Alvorlig blødning blev observeret hos 1 patient (0,2%) af hver gruppe.
Behandling af patienter med spontan symptomatisk akut overfladisk venetrombose uden samtidig dyb venetrombose (DVT)
Et randomiseret, dobbeltblindet klinisk forsøg (CALISTO) omfattede 3002 patienter med spontan, akut, symptomatisk og isoleret overfladisk venetrombose i underekstremiteterne, mindst 5 cm i længden, bekræftet af kompressionsultralyd (CUS). Patienter blev ikke inkluderet, hvis de havde samtidig DVT eller overfladisk venetrombose inden for 3 cm fra sapheno-femoral junction. Patienter blev udelukket, hvis de havde alvorlig leverinsufficiens, alvorlig nyreinsufficiens (kreatininclearance
Patienter blev randomiseret til at modtage fondaparinux 2,5 mg én gang dagligt eller placebo i 45 dage ud over strømper, smertestillende midler og / eller topiske ikke-steroide antiinflammatoriske lægemidler (NSAID'er). Opfølgningen fortsatte indtil dag 77. Studiepopulationen var 64% kvinder, med en medianalder på 58 år, 4,4% havde kreatininclearance.
Det primære effektresultat, et sammensat resultat af symptomatisk PE, symptomatisk DVT, forlængelse af symptomatisk overfladisk venøs trombose, gentagelse af symptomatisk overfladisk venøs trombose eller død på dag 47, blev signifikant reduceret med 5,9% hos patienter i placebogruppen. 0,9% hos dem, der modtog fondaparinux 2,5 mg (relativ risikoreduktion: 85,2%; 95% CI, 73,7% til 91,7% [p
Forekomsten af hver tromboembolisk komponent i det primære resultat var også signifikant reduceret hos fondaparinux -patienter som beskrevet nedenfor: symptomatisk PE [0 (0%) vs 5 (0,3%) (p = 0,031)], symptomatisk DVT [3 (0,2%) vs 18 (1,2%); relativ risikoreduktion 83,4% (s
Dødeligheden var lav og lignende mellem behandlingsgrupperne med 2 (0,1%) dødsfald i fondaparinux -gruppen imod 1 (0,1%) død i placebogruppen.
Effektiviteten blev opretholdt til og med dag 77 og var konsistent på tværs af alle foruddefinerede undergrupper, herunder patienter med åreknuder og patienter med overfladisk venetrombose placeret under knæet.
Større blødninger under behandlingen forekom hos 1 (0,1%) patient på fondaparinux og hos 1 (0,1%) patient på placebo. Klinisk relevant ikke-større blødning forekom hos 5 (0,3%) patienter på fondaparinux og hos 8 (0,5%) patienter på placebo.
05.2 Farmakokinetiske egenskaber
Absorption
Efter subkutan administration absorberes fondaparinux fuldstændigt og hurtigt (100% absolut biotilgængelighed). Efter en enkelt subkutan injektion af fondaparinux 2,5 mg til raske unge personer opnås maksimal plasmakoncentration (gennemsnitlig C = 0,34 mg / l) 2 timer efter administration. Plasmakoncentrationer svarende til halvdelen af de gennemsnitlige Cmax -værdier nås 25 minutter efter administration.
Fondaparinux farmakokinetik er lineær over et dosisinterval på 2 til 8 mg subkutant hos raske ældre personer. Efter én gang daglig dosering opnås steady-state plasmaniveauer 3 til 4 dage senere med en 1,3 gange stigning i Cmax og AUC.
Middelværdien (CV%) af de estimerede steady state -parametre for fondaparinux hos patienter med hofteerstatningskirurgi, der fik fondaparinux 2,5 mg én gang dagligt, er: Cmax (mg / l) - 0,39 (31%), Tmax (t) - 2,8 (18% ) og Cmin (mg / l) - 0,14 (56%). Hos patienter med hoftebrud forbundet med alderdom er fondaparinux plasmakoncentrationer ved steady state: Cmax (mg / l) - 0,50 (32%), Cmin (mg / l / l) - 0,19 (58%).
Fordeling
Distributionsvolumen for fondaparinux er begrænset (7 - 11 liter). In vitro, fondaparinux er stærkt og specifikt bundet til antithrombinprotein med en dosisafhængig plasmakoncentrationsbinding (98,6% til 97,0% over et koncentrationsinterval på 0,5 til 2 mg / l). Fondaparinux binder ikke signifikant til andre plasmaproteiner, herunder trombocytfaktor 4 (PF4).
Da fondaparinux ikke binder signifikant til andre plasmaproteiner end ATIII, forventes der ingen interaktion med andre lægemidler ved at ændre proteinbinding.
Biotransformation
Selvom det ikke er fuldstændigt evalueret, er der ingen beviser for fondaparinux -metabolisme og især for dannelsen af aktive metabolitter.
Fondaparinux hæmmer ikke in vitro CYP450 -systemet (CYP1A2, CYP2A6, CYP2C9, CYP2C19, CYP2D6, CYP2E1 eller CYP3A4). Derfor forventes det ikke, at fondaparinux vil interagere in vivo med andre lægemidler ved at hæmme CYP-medieret metabolisme.
Eliminering
Elimineringshalveringstiden (t½) er cirka 17 timer hos raske unge forsøgspersoner og cirka 21 timer hos raske ældre. Fondaparinux udskilles med 64 til 77% af nyrerne som uændret forbindelse.
Særlige kategorier af patienter:
Pædiatrisk population - Fondaparinux er ikke blevet undersøgt i denne patientklasse til forebyggelse af VTE eller til behandling af overfladisk venetrombose.
Ældre patienter - Nyrefunktionen kan falde med alderen, og derfor kan eliminationskapaciteten af fondaparinux reduceres hos ældre. Hos patienter> 75 år, der blev opereret, var den estimerede plasmaclearance 1,2 til 1,4 gange lavere end hos patienter med alder
Nyresvigt - Sammenlignet med patienter med normal nyrefunktion (kreatininclearance> 80 ml / min) er plasmaclearance 1,2 til 1,4 gange lavere hos patienter med let nedsat nyrefunktion (kreatininclearance 50 til 80 ml / min) og i gennemsnit 2 gange lavere hos patienter med moderat nedsat nyrefunktion (kreatininclearance 30 til 50 ml / min). Ved alvorlig nyreinsufficiens (kreatininclearance
Køn - Der var ingen forskel mellem kønnene efter justering for kropsvægt.
Race - Farmakokinetiske forskelle på grund af race er ikke undersøgt prospektivt. Undersøgelser udført på asiatiske (japanske) raske forsøgspersoner afslørede imidlertid ikke en anden farmakokinetisk profil sammenlignet med kaukasiske raske forsøgspersoner. På samme måde blev der ikke observeret forskelle i plasmaclearance mellem sorte og kaukasiske patienter, der gennemgik ortopædkirurgi.
Kropsvægt - Plasmaclearance for fondaparinux stiger med kropsvægt (9% stigning pr. 10 kg).
Leverinsufficiens - Efter en enkelt subkutan dosis fondaparinux hos personer med moderat nedsat leverfunktion (Child-Pugh kategori B) blev den samlede (dvs. både bundne og ubundne) Cmax og AUC reduceret med henholdsvis 22% og 39% i sammenligning med forsøgspersoner med normal leverfunktion. De lavere plasmakoncentrationer af fondaparinux er blevet tilskrevet den reducerede binding til ATIII, som igen er afhængig af de lavere plasmakoncentrationer af ATIII hos personer med leverinsufficiens, hvilket derfor resulterer i en stigning i renal clearance af fondaparinux. frie fondaparinux -koncentrationer forventes at forblive uændrede hos patienter med let eller moderat nedsat leverfunktion, og derfor er det ikke nødvendigt at justere dosis baseret på farmakokinetik.
05.3 Prækliniske sikkerhedsdata
Ikke-kliniske data afslører ingen særlig fare for mennesker baseret på konventionelle undersøgelser af sikkerhedsfarmakologi, toksicitet ved gentagne doser og toksicitet Dyrestudier er utilstrækkelige med hensyn til reproduktionstoksiske virkninger på grund af begrænset eksponering.
06.0 LÆGEMIDDELOPLYSNINGER
06.1 Hjælpestoffer
Natriumchlorid
Vand til injektionsvæsker
Saltsyre
Natriumhydroxid
06.2 Uforenelighed
I mangel af kompatibilitetsundersøgelser må dette lægemiddel ikke blandes med andre lægemidler.
06.3 Gyldighedsperiode
3 år.
06.4 Særlige opbevaringsforhold
Opbevares ved temperaturer under 25 ° C. Må ikke fryses.
06.5 Den umiddelbare emballages art og emballagens indhold
Type I -glas (1 ml) udstyret med en 27 gauge x 12,7 mm nål og låst af et bromobutyl- eller chlorbutylelastomerstemplelåsningssystem.
Arixtra fås i pakninger med 2, 7, 10 og 20 fyldte sprøjter. Der er to typer sprøjter:
• sprøjte med gult stempel og med et automatisk sikkerhedssystem
• sprøjte med et gult stempel og et manuelt sikkerhedssystem.
Ikke alle pakningsstørrelser er nødvendigvis markedsført.
06.6 Brugsanvisning og håndtering
Den subkutane injektion administreres som med en konventionel sprøjte.
Parenterale opløsninger bør undersøges visuelt før administration for unormale partikler og farvning.
Instruktioner til selvadministration findes i indlægssedlen.
Nålbeskyttelsessystemet til Arixtra fyldte sprøjter er designet med et sikkerhedssystem til beskyttelse mod utilsigtede nålepinde efter injektion.
Ubrugt medicin og affald fra denne medicin skal bortskaffes i overensstemmelse med lokale regler.
07.0 INDEHAVER AF MARKEDSFØRINGSTILLADELSE
Aspen Pharma Trading Limited
3016 Lake Drive
Citywest Business Campus
Dublin 24
Irland
08.0 MARKEDSFØRINGSTILLADELSESNUMMER
EU / 1/02/206 / 005-008
035606060
035606072
EU/1/02/206/024
EU/1/02/206/025
035606223
EU/1/02/206/026
09.0 DATO FOR FØRSTE TILLADELSE ELLER FORNYELSE AF TILLADELSEN
Dato for første godkendelse: 21. marts 2002
Dato for sidste fornyelse: 21. marts 2007
10.0 DATO FOR REVISION AF TEKSTEN
D.CCE august 2014