Aktive ingredienser: Strontium (strontiumranelat)
PROTELOS 2 g granulat til oral suspension
Hvorfor bruges Protelos? Hvad er det for?
PROTELOS er en medicin, der bruges til behandling af alvorlig osteoporose:
- hos postmenopausale kvinder
- hos voksne mænd
med stor risiko for brud, for hvilke det ikke er muligt at ty til alternative behandlinger. Hos postmenopausale kvinder reducerer strontiumranelat risikoen for ryg- og hoftebrud.
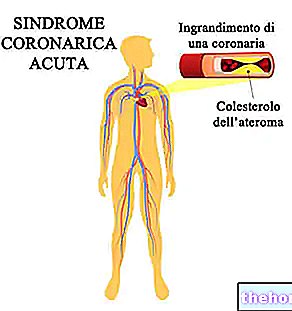
Osteoporose
Kroppen ødelægger hele tiden gammel knogle og danner ny knogle.I tilfælde af knogleskørhed ødelægger kroppen mere knogle end der dannes, så der gradvist opstår knogletab og knoglerne bliver tyndere og mere skrøbelige.Det forekommer især hos kvinder efter overgangsalderen.
Mange mennesker med osteoporose har ingen symptomer, og det er muligt ikke engang at vide, at du har osteoporose. Imidlertid disponerer osteoporose for brud (knækkede knogler) især i rygsøjlen, hofter og håndled.
Sådan fungerer PROTELOS
PROTELOS, der indeholder det aktive stof strontiumranelat, tilhører en gruppe lægemidler, der bruges til behandling af knoglesygdomme. PROTELOS reducerer knogledestruktion og stimulerer knoglerekonstruktion og reducerer derved risikoen for brud. Den nye knogle er dannet af normal kvalitet.
Kontraindikationer Når Protelos ikke bør bruges
Tag ikke PROTELOS
- hvis du er allergisk over for strontiumranelat eller et af de øvrige indholdsstoffer i PROTELOS (angivet i afsnit 6).
- hvis du har eller nogensinde har haft en blodprop (f.eks. påvirker blodkarrene i benet eller lungerne).
- hvis du er immobiliseret permanent eller i en bestemt periode, f.eks. hvis du sidder i kørestol, eller hvis du er sengeliggende, eller hvis du skal opereres, eller hvis du er i postoperativ restitution. Risikoen for venøs trombose (trombose i benet eller lungen) kan være højere ved langvarig immobilisering.
- hvis du har kendt iskæmisk hjertesygdom eller cerebrovaskulær sygdom, f.eks. hvis du er blevet diagnosticeret med et hjerteanfald, slagtilfælde eller forbigående iskæmisk anfald (midlertidig reduktion i blodgennemstrømningen til hjernen; også kendt som et "mini-slagtilfælde"), angina eller en blokering af blodkar i hjertet eller hjernen .
- hvis du har eller har haft problemer med din blodcirkulation (perifer arteriel sygdom), eller hvis du har været opereret i arterierne i dine ben.
- hvis du har forhøjet blodtryk, som ikke kontrolleres af behandlingen.
Forholdsregler ved brug Hvad du skal vide, før du tager Protelos
Tal med din læge eller apotek, før du tager PROTELOS:
- hvis du har risiko for hjertesygdomme dette omfatter forhøjet blodtryk, højt kolesteroltal, diabetes, rygning
- hvis du har risiko for trombose
- hvis du har en alvorlig nyresygdom.
Din læge vil evaluere tilstanden i dit hjerte og dine blodkar med jævne mellemrum, normalt hver 6.-12. Måned, i hele behandlingsperioden med PROTELOS.
Hvis du under behandlingen får en allergisk reaktion (såsom hævelse af ansigt, tunge eller hals, åndedrætsbesvær eller synke, hududslæt), skal du straks stoppe med at tage PROTELOS og kontakte din læge (se afsnit 4). Potentielt livstruende hududslæt (Stevens-Johnsons syndrom (SJS), toksisk epidermal nekrolyse og alvorlige overfølsomhedsreaktioner (DRESS)) er blevet rapporteret under brug af PROTELOS. Den største risiko for forekomst af alvorlige hudreaktioner er inden for de første ugers behandling for Stevens-Johnsons syndrom og toksisk epidermal nekrolyse og normalt omkring 3-6 uger til DRESS.Hvis du får udslæt eller alvorlige hudsymptomer (se afsnit 4), skal du stoppe med at tage PROTELOS, straks kontakte din læge. Og fortæl din læge, at du tager det Hvis du har oplevet Stevens-Johnsons syndrom, toksisk epidermal nekrolyse eller DRESS under brug af PROTELOS, bør du aldrig genstarte behandlingen med PROTELOS. Hvis du er af asiatisk afstamning, skal du tale med din læge, før du tager PROTELOS, da du kan have en højere risiko for hudreaktioner.
Børn og unge
PROTELOS er ikke indiceret til brug hos børn og unge (under 18 år).
Interaktioner Hvilke lægemidler eller fødevarer kan ændre virkningen af Protelos
Andre lægemidler og PROTELOS
Fortæl det til din læge eller apotek, hvis du tager anden medicin eller har brugt det for nylig.
Stop med at tage PROTELOS, hvis du skal tage orale tetracycliner, f.eks. Doxycyclin eller quinoloner, f.eks. Ciprofloxacin (to typer antibiotika). Du kan genstarte PROTELOS, når du er færdig med at tage disse antibiotika. Spørg din læge eller apotek, hvis du er i tvivl. du tager medicin, der indeholder calcium, skal der gå mindst 2 timer, før du tager PROTELOS.
Hvis du tager antacida (medicin mod halsbrand), skal du tage dem mindst 2 timer efter at have taget PROTELOS. Hvis dette ikke er muligt, er det acceptabelt at tage begge lægemidler sammen.
Hvis det er nødvendigt at teste calciumniveauet i blodet eller urinen, skal du informere laboratoriet om, at du tager PROTELOS, da det kan forstyrre nogle testmetoder.
Sammen med mad og drikke
Fødevarer, mælk og dets derivater reducerer absorptionen af strontiumranelat. Det anbefales at tage PROTELOS i intervallet mellem måltiderne, helst ved sengetid, mindst to timer efter mad, mælk og mælkederivater eller kalktilskud.
Advarsler Det er vigtigt at vide, at:
Graviditet og amning
Tag ikke PROTELOS under graviditet eller amning. I tilfælde af utilsigtet indtagelse under graviditet eller amning, skal du straks stoppe med at tage medicinen og informere din læge.
Kørsel og brug af maskiner
PROTELOS påvirker sandsynligvis ikke din evne til at føre motorkøretøj eller betjene maskiner.
PROTELOS indeholder aspartam (E951)
Hvis du har phenylketonuri (en sjælden nedarvet metabolisme), skal du kontakte din læge, før du begynder at tage denne medicin.
Dosis, metode og administrationstidspunkt Sådan bruges Protelos: Dosering
Behandlingen bør kun startes af en læge med erfaring i behandling af osteoporose.
Tag altid dette lægemiddel nøjagtigt efter lægens eller apotekspersonalets anvisning. Hvis du er i tvivl, skal du kontakte din læge eller apotek.
PROTELOS er til oral brug. Den anbefalede dosis er en 2 g pose om dagen.
Det anbefales at tage PROTELOS ved sengetid, helst mindst 2 timer efter middagen. Du kan også gå i seng umiddelbart efter at have taget PROTELOS, hvis du ønsker det.
Tag granulatet i poserne efter at have sat det i suspension i et glas, der indeholder mindst 30 ml vand (ca. en tredjedel af et standardglas) (se instruktionerne herunder). PROTELOS kan interagere med mælk og dets derivater; derfor er det Det er vigtigt, at PROTELOS kun blandes med vand for at sikre, at medicinen virker korrekt.
- Hæld granulatet fra posen i et glas;
- Tilsæt vand;
- Rør rundt, indtil granulatet er helt spredt i vandet.
Drik med det samme. Lad ikke mere end 24 timer gå, før du drikker suspensionen. Hvis du af en eller anden grund ikke kan tage medicinen med det samme, skal du huske at blande den igen, før du drikker.
Din læge kan råde dig til at tage calcium- og D -vitamintilskud ud over PROTELOS. Tag ikke calciumtilskud ved sengetid, samtidig med PROTELOS.
Din læge vil fortælle dig, hvor længe du skal fortsætte med at tage PROTELOS. Behandling af osteoporose tager normalt lang tid.Det er vigtigt at fortsætte med at tage PROTELOS, så længe din læge foreskriver det.
Overdosering Hvad skal man gøre, hvis man har taget for meget Protelos
Hvis du har taget for mange PROTELOS
Hvis du tager flere PROTELOS -poser, end din læge har ordineret, skal du fortælle det til din læge eller apotek. De kan råde dig til at drikke mælk eller tage antacida for at reducere absorptionen af den aktive ingrediens.
Hvis du har glemt at tage PROTELOS
Tag ikke en dobbelt dosis for at kompensere for en glemt dosis. Tag blot den næste dosis på det aftalte tidspunkt.
Hvis du holder op med at tage PROTELOS
Det er vigtigt at fortsætte med at tage PROTELOS, så længe din læge har ordineret det. PROTELOS kan kun behandle alvorlig osteoporose, hvis det tages kontinuerligt. Spørg din læge eller apotek, hvis du har yderligere spørgsmål om brugen af denne medicin.
Bivirkninger Hvad er bivirkningerne af Protelos
Ligesom al anden medicin kan denne medicin forårsage bivirkninger, men ikke alle får bivirkninger.
Stop med at tage PROTELOS, og fortæl det straks til din læge, hvis en af følgende bivirkninger opstår:
Almindelig (kan forekomme hos op til 1 ud af 10 patienter):
- Hjerteanfald: Pludselige, klemmende smerter i brystet, der kan strække sig til venstre arm, kæbe, mave, ryg og / eller skuldre. Andre symptomer kan være: kvalme / opkastning, svedtendens, åndenød, hjertebanken, (ekstrem) træthed og / eller svimmelhed. Hos patienter med høj risiko for hjertesygdomme kan der forekomme et hjerteanfald med en almindelig hyppighed. Hvis du er en højrisikopatient, vil din læge ikke ordinere PROTELOS til dig.
- Blodpropper i venerne (trombose): smerter, rødme, hævelse af benene, pludselige brystsmerter eller åndedrætsbesvær.
Sjælden (kan forekomme hos op til 1 ud af 1.000 patienter):
- Tegn på alvorlige overfølsomhedsreaktioner (DRESS): i første omgang som influenzalignende symptomer og udslæt i ansigtet, derefter forlænget udslæt med høj temperatur (ikke almindelig), stigning i leverenzymniveauer fundet i blodprøver (ikke almindelig), stigning i en bestemt type af hvide blodlegemer (eosinofili) (sjælden) og forstørrede lymfeknuder (ikke almindelig).
Meget sjælden (kan forekomme hos op til 1 ud af 10.000 patienter):
- Tegn på potentielt livstruende hududslæt (Stevens-Johnsons syndrom, toksisk epidermal nekrolyse): I første omgang som rødlige mållignende pletter eller cirkulære pletter ofte med centrale blærer på bagagerummet. Yderligere tegn kan omfatte sårdannelse i mund, hals, næse, kønsorganer og konjunktivitis (hævede og røde øjne). Disse potentielt livstruende hududslæt ledsages ofte af influenzalignende symptomer. Udslæt kan udvikle sig til blærer over hele kroppen eller afskalning af huden.
Andre mulige bivirkninger
Meget almindelig (kan forekomme hos mere end 1 ud af 10 patienter):
Kløe, nældefeber, hududslæt, angioødem (såsom hævelse af ansigt, tunge eller hals, åndedræts- eller synkebesvær), smerter i knogler, lemmer, muskler og / eller led, muskelkramper.
almindelige
Opkastning, mavesmerter, tilbagesvaling, fordøjelsesbesvær, forstoppelse, flatulens, søvnbesvær, leverbetændelse (hepatitis), hævelse af lemmerne, bronkial hyperreaktivitet (symptomer inkluderer hvæsen, åndenød og hoste), øget niveau af enzymmuskel (kreatinfosfokinase). Kvalme, diarré, hovedpine, eksem, hukommelsessvigt, besvimelse, prikken, svimmelhed, svimmelhed.Disse virkninger er imidlertid milde og forbigående og kræver normalt ikke afbrydelse af behandlingen. Fortæl det til din læge, hvis nogen af disse bivirkninger bliver generende eller vedvarende.
Ikke almindelig (kan forekomme hos op til 1 ud af 100 patienter):
(Kramper, irritation af mundslimhinden (såsom sår i munden og betændelse i tandkødet), hårtab, forvirring, kvalme, tør mund, hudirritation.
Sjælden:
Nedsat produktion af blodlegemer i knoglemarven. Hvis du har stoppet behandlingen på grund af overfølsomhedsreaktioner, bør du ikke genstarte PROTELOS.
Indberetning af bivirkninger
Tal med din læge eller apotek, hvis du får bivirkninger, herunder mulige bivirkninger, som ikke er nævnt i denne indlægsseddel. Du kan også indberette bivirkninger direkte via det nationale rapporteringssystem anført i tillæg V. Ved at rapportere bivirkninger kan du hjælpe med at give mere information om sikkerheden ved dette lægemiddel.
Udløb og opbevaring
Opbevar denne medicin utilgængeligt for børn.
Brug ikke dette lægemiddel efter den udløbsdato, der står på kartonen og posen efter EXP. Udløbsdatoen refererer til den sidste dag i den pågældende måned.
Denne medicin kræver ingen særlige opbevaringsbetingelser.
Efter rekonstituering i vand er suspensionen stabil i 24 timer. Det anbefales dog at drikke suspensionen umiddelbart efter tilberedning (se afsnit 3).
Smid ikke medicin via spildevand eller husholdningsaffald. Spørg din apotek om, hvordan du skal smide medicin, du ikke længere bruger. Dette vil hjælpe med at beskytte miljøet.
Sammensætning og farmaceutisk form
Hvad PROTELOS indeholder
- Den aktive ingrediens er strontiumranelat. Hver pose indeholder 2 g strontiumranelat.
- Øvrige indholdsstoffer er aspartam (E 951), maltodextrin, mannitol (E 421).
Beskrivelse af hvordan PROTELOS ser ud og pakningens indhold
PROTELOS fås i poser indeholdende et gult granulat til oral suspension. PROTELOS leveres i pakker med 7, 14, 28, 56, 84 eller 100 breve. Ikke alle pakningsstørrelser er nødvendigvis markedsført.
Indlægsseddel: AIFA (Italian Medicines Agency). Indhold offentliggjort i januar 2016. De foreliggende oplysninger er muligvis ikke opdaterede.
For at få adgang til den mest opdaterede version er det tilrådeligt at få adgang til webstedet AIFA (Italian Medicines Agency). Ansvarsfraskrivelse og nyttige oplysninger.
01.0 LÆGEMIDLETS NAVN
PROTELOS 2 G
▼ Lægemiddel underlagt yderligere overvågning. Dette vil muliggøre hurtig identifikation af nye sikkerhedsoplysninger. Sundhedspersonale bedes rapportere alle formodede bivirkninger Se afsnit 4.8 for information om, hvordan man rapporterer bivirkninger.
02.0 KVALITATIV OG KVANTITATIV SAMMENSÆTNING
Hver pose indeholder 2 g strontiumranelat.
Hjælpestof med kendt virkning: hver pose indeholder også 20 mg aspartam (E 951).
Den fulde liste over hjælpestoffer findes i afsnit 6.1.
03.0 LÆGEMIDDELFORM
Granulat til oral suspension.
Gul granulat.
04.0 KLINISKE OPLYSNINGER
04.1 Terapeutiske indikationer
Behandling af alvorlig osteoporose:
- hos postmenopausale kvinder
- hos voksne mænd
med stor risiko for brud, for hvilke behandling med andre lægemidler, der er godkendt til behandling af osteoporose, ikke er mulig på grund af f.eks. kontraindikationer eller intolerance.
Strontiumranelat reducerer risikoen for hvirvel- og hoftebrud hos postmenopausale kvinder (se pkt.5.1).
Beslutningen om at ordinere strontiumranelat bør baseres på en vurdering af den enkelte patients samlede risici (se afsnit 4.3 og 4.4).
04.2 Dosering og indgivelsesmåde
Behandlingen bør kun startes af en læge med erfaring i behandling af osteoporose.
Dosering
Den anbefalede dosis er en 2 g pose én gang dagligt til oral administration.
På grund af tilstanden, der behandles, er strontiumranelat beregnet til langvarig brug.
Absorption af strontiumranelat reduceres fra mad, mælk og dets derivater, og derfor bør PROTELOS administreres mellem måltiderne. På grund af dets langsomme absorption skal PROTELOS tages ved sengetid, helst mindst to timer efter et måltid (se afsnit 4.5 og 5.2).
Patienter, der behandles med strontiumranelat, bør tage D -vitamin og calciumtilskud, hvis deres kostindtag er utilstrækkeligt.
Ældre patienter
Effekten og sikkerheden af strontiumranelat er blevet påvist i en stor prøve af voksne mænd og postmenopausale kvinder i alle aldre (op til 100 år ved inklusion) med osteoporose. Ingen dosisjustering er nødvendig i forhold til alder.
Patienter med nyreinsufficiens
Strontiumranelat anbefales ikke til patienter med svært nedsat nyrefunktion (kreatininclearance mindre end 30 ml / min) (se pkt. 4.4 og 5.2). Ingen dosisjustering er nødvendig hos patienter med let til moderat nedsat nyrefunktion (kreatininclearance 30-70 ml / min) (se pkt. 4.4 og 5.2).
Patienter med leverinsufficiens
Dosisjustering er ikke påkrævet hos patienter med leverinsufficiens (se pkt. 5.2).
Pædiatrisk population
Sikkerheden og effekten af PROTELOS hos børn under 18 år er ikke fastslået.
Ingen data er tilgængelige.
Indgivelsesmåde
Til oral brug.
Posenes granulat skal tages efter suspension i et glas, der indeholder mindst 30 ml vand (ca. en tredjedel af et normalt glas).
Selvom brugsundersøgelser har vist, at strontiumranelat forbliver stabil i suspension i 24 timer efter tilberedning, bør suspensionen drikkes umiddelbart efter tilberedning.
04.3 Kontraindikationer
- Overfølsomhed over for det aktive stof eller over for et eller flere af hjælpestofferne anført i pkt.6.1.
- Aktuel eller tidligere venøs tromboemboli (VTE), herunder dyb venetrombose og lungeemboli.
- Midlertidig eller permanent immobilisering, f.eks. På grund af kirurgi eller et længere ophold i sengen.
- Kendt, nuværende eller tidligere iskæmisk hjertesygdom, perifer arteriel sygdom og / eller cerebrovaskulær sygdom.
- Ukontrolleret hypertension.
04.4 Særlige advarsler og passende forholdsregler ved brug
Iskæmiske hjertehændelser
I en samlet analyse af placebokontrollerede randomiserede kliniske forsøg med postmenopausale osteoporotiske patienter blev der observeret en signifikant stigning i myokardieinfarkt hos patienter behandlet med PROTELOS sammenlignet med dem, der blev behandlet med placebo (se pkt. 4.8).
Patienter bør vurderes for kardiovaskulær risiko, inden behandlingen påbegyndes.
Patienter med betydelige risikofaktorer for kardiovaskulære hændelser (f.eks. Hypertension, hyperlipidæmi, diabetes mellitus, rygning) bør kun behandles med strontiumranelat efter grundig overvejelse (se pkt. 4.3 og 4.8).
Under behandling med PROTELOS bør disse kardiovaskulære risici overvåges med jævne mellemrum, generelt hver 6.-12. Måned.
Behandlingen bør afbrydes, hvis patienten udvikler iskæmisk hjertesygdom, perifer arteriel sygdom, cerebrovaskulær sygdom eller hvis hypertension ikke er kontrolleret (se pkt. 4.3).
Venøs tromboemboli
I fase III placebokontrollerede undersøgelser var behandling med strontiumranelat forbundet med en øget årlig forekomst af venøs tromboemboli (VTE), herunder lungeemboli (se pkt. 4.8). Årsagen til denne stigning er ukendt. PROTELOS er kontraindiceret til patienter med tidligere venøs tromboemboli (se pkt. 4.3) og bør anvendes med forsigtighed til patienter med risiko for VTE.
Under behandlingen af patienter over 80 år med risiko for VTE bør behovet for at fortsætte behandlingen med PROTELOS revurderes. Behandling med PROTELOS bør afbrydes hurtigst muligt i tilfælde af sygdom eller tilstand, der fører til immobilisering (se afsnit 4.3), og passende forebyggende foranstaltninger bør træffes. Terapien bør ikke genoptages, før tilstanden, der har ført til immobilisering, ikke er løst og patienten er helt mobil. Når der opstår en VTE, skal PROTELOS afbrydes.
Anvendelse til patienter med nyreinsufficiens
I mangel af knoglesikkerhedsdata hos patienter med alvorlig nyreinsufficiens, der modtager strontiumranelat, anbefales PROTELOS ikke til patienter med en kreatininclearance under 30 ml / min. (se afsnit 5.2). I overensstemmelse med god klinisk praksis anbefales periodisk monitorering af nyrefunktionen hos patienter med kronisk nyresvigt. Fortsættelse af PROTELOS -behandling hos patienter, der udvikler alvorlig nyreinsufficiens, bør vurderes individuelt.
Hudreaktioner
Livstruende hudreaktioner (Stevens-Johnsons syndrom (SJS), toksisk epidermal nekrolyse (NET) og lægemiddeludslæt med eosinofili og systemiske symptomer (DRESS)) er blevet rapporteret under brug af PROTELOS.
Patienter skal informeres om tegn og symptomer og overvåges nøje for hudreaktioner. Den største risiko for forekomst af SJS eller NET er inden for de første par uger af behandlingen og inden for 3-6 uger for DRESS.
Hvis tegn og symptomer på SJS eller NET (f.eks. Progressivt hududslæt ofte med blærer og slimhinde læsioner) eller KJOLE (f.eks. Udslæt, feber, eosinofili og systemisk involvering (f.eks. Adenopati, hepatitis, nefropati og lungesygdom) forekommer interstitiel), PROTELOS behandling bør stoppes med det samme.
De bedste resultater i forvaltningen af SJS, NET eller DRESS opnås efter en tidlig diagnose og øjeblikkelig seponering af ethvert mistænkt lægemiddel. Tidlig seponering af behandlingen er forbundet med en bedre prognose. Det kliniske billede af DRESS forsvandt i de fleste tilfælde med afbrydelse af PROTELOS -behandlingen og med påbegyndelse af kortikosteroidbehandling, når det er nødvendigt. Gendannelsen kan være langsom, og i nogle tilfælde er der rapporteret om tilbagefald af syndromet efter afbrydelse af kortikosteroidbehandling.
Hos patienter, der har udviklet SJS, NET eller DRESS ved brug af PROTELOS, bør behandling med PROTELOS ikke længere genstartes.
En højere, men stadig sjælden, forekomst af overfølsomhedsreaktioner inklusive hududslæt, SJS eller NET er blevet rapporteret hos patienter af asiatisk afstamning.
Interaktioner med laboratorietest
Strontium forstyrrer kolorimetriske metoder til bestemmelse af blod- og urinkoncentrationer af calcium. Derfor skal der i klinisk praksis anvendes induktivt koblede plasma -atomemissionsspektrometri eller atomabsorptionsspektrometri -metoder til at sikre nøjagtig vurdering af blod- og urinkalciumkoncentrationer.
Hjælpestof
PROTELOS indeholder aspartam, en kilde til phenylalanin, som kan være farlig for patienter med phenylketonuri.
04.5 Interaktioner med andre lægemidler og andre former for interaktion
Fødevarer, mælk og dets derivater og medicinske specialiteter, der indeholder calcium, kan reducere biotilgængeligheden af strontiumranelat med cirka 60 - 70%. Derfor bør administrationen af PROTELOS og disse produkter adskilles med mindst to timer (se afsnit 4.2 og 5.2).
Da divalente kationer kan danne et dårligt absorberbart kompleks med orale tetracycliner (f.eks. Doxycyclin) og quinolonantibiotika (f.eks. Ciprofloxacin) på mave-tarm-niveau, anbefales samtidig administration af strontiumranelat med disse lægemidler ikke. Som en sikkerhedsforanstaltning bør PROTELOS seponeres under behandling med orale tetracykliner eller quinolonantibiotika.
Et klinisk studie in vivo om lægemiddelinteraktioner har vist, at indtag af aluminium og magnesiumhydroxider i de to timer før eller samtidig med strontiumranelat forårsagede et let fald i absorptionen af strontiumranelat (20-25% fald i AUC), mens absorptionen forblev praktisk talt uændret, da antacida blev administreret to timer efter strontiumranelatet.Det foretrækkes derfor at tage antacida mindst to timer efter indtagelse af PROTELOS. Men da det anbefales, at PROTELOS tages ved sengetid, når denne doseringsplan ikke er relevant, forbliver samtidig indtag acceptabelt.
Der blev ikke observeret nogen interaktion med oral vitamin D -tilskud.
I kliniske forsøg er der ikke påvist nogen klinisk interaktion eller en signifikant stigning i strontiumniveauer i blod med lægemidler, der i den nuværende praksis sædvanligvis ordineres samtidigt med PROTELOS, herunder: ikke-steroide antiinflammatoriske lægemidler (herunder acetylsalicylsyre) , anilider (såsom paracetamol), H2-blokkere og protonpumpehæmmere, diuretika, digoxin og hjerteglykosider, organiske nitrater og andre vasodilatatorer til hjertesygdomme, calciumkanalblokkere, betablokkere, ACE-hæmmere, angiotensin II-antagonister, selektive beta-2- adrenerge receptoragonister, orale antikoagulantia, trombocytaggregationshæmmere, statiner, fibrater og benzodiazepinderivater.
04.6 Graviditet og amning
Graviditet
Data om brugen af strontiumranelat til gravide er ikke tilgængelige Dyrestudier har ved høje doser vist reversible knogleeffekter hos afkom af rotter og kaniner, der blev behandlet under graviditet (se pkt. 5.3) Hvis PROTELOS tages utilsigtet under graviditeten, behandling bør afbrydes.
Fodringstid
Fysisk-kemiske data tyder på udskillelse af strontiumranelat i modermælk.PROTELOS bør ikke anvendes under amning.
Fertilitet
Der blev ikke observeret nogen effekt på fertiliteten hos mænd og kvinder i dyreforsøg.
04.7 Virkninger på evnen til at føre motorkøretøj og betjene maskiner
Strontiumranelat har ingen eller ubetydelig indflydelse på evnen til at føre motorkøretøj eller betjene maskiner.
04.8 Bivirkninger
Resumé af sikkerhedsprofilen
PROTELOS er blevet undersøgt i kliniske forsøg med ca. 8.000 mennesker. Langsigtet sikkerhed blev evalueret i fase III-undersøgelser hos postmenopausale kvinder med osteoporose behandlet i op til 60 måneder med strontiumranelat 2 g / dag (n = 3.352) eller placebo (n = 3.317). Middelalderen på inklusionstidspunktet var 75 år, og 23% af de tilmeldte patienter var mellem 80 og 100 år gamle.
I en samlet analyse af randomiserede placebokontrollerede forsøg med postmenopausale osteoporotiske patienter var de mest almindelige bivirkninger kvalme og diarré, generelt rapporteret ved behandlingsstart, uden nogen mærkbar forskel mellem grupper i de senere stadier. Afbrydelse af behandlingen skyldtes hovedsageligt kvalme Der var ingen forskel i karakteren af bivirkninger mellem behandlingsgrupperne, uanset om patienterne var under eller over 80 år på inklusionstidspunktet.
Tabel over bivirkninger
Følgende bivirkninger er blevet rapporteret under kliniske forsøg og / eller under markedsføring af strontiumranelat efter markedsføring. Bivirkninger er anført nedenfor ved hjælp af følgende konvention: meget almindelig (≥1 / 10); almindelig (≥1 / 100 op til
§ Hyppigheden i kliniske forsøg var ens i lægemiddelgruppen og placebogruppen.
* Rapporteret som sjælden i asiatiske lande.
# For bivirkninger, der ikke er observeret i kliniske forsøg, er den øvre grænse for 95% konfidensintervallet ikke større end 3 / X, hvor X repræsenterer den samlede stikprøvestørrelse fra alle kliniske forsøg og relevante undersøgelser.
en muskuloskeletal fraktion> 3 gange den øvre grænse for normalområdet. I de fleste tilfælde normaliserede disse værdier sig spontant uden ændringer i terapien.
Beskrivelse af udvalgte bivirkninger
Venøs tromboemboli
I fase III -undersøgelser var den årlige forekomst af venøs tromboemboli (VTE) hændelser observeret over 5 år cirka 0,7% med en relativ risiko på 1,4 (95% CI = [1,0; 2, 0]) hos patienter behandlet med strontiumranelat versus placebo (se pkt. 4.4).
Myokardieinfarkt
I en samlet analyse af placebokontrollerede randomiserede kliniske forsøg med postmenopausale osteoporotiske patienter blev der observeret en signifikant stigning i myokardieinfarkt hos patienter behandlet med strontiumranelat sammenlignet med patienter, der fik placebo (1,7% sammenlignet med 1,1%), med en relativ risiko for 1,6 (95% CI = [1,07; 2,38]).
Indberetning af formodede bivirkninger
Rapportering af formodede bivirkninger, der opstår efter godkendelse af lægemidlet, er vigtig, da det muliggør kontinuerlig overvågning af lægemidlets fordel / risiko -balance.Professionelle sundhedspersonale anmodes om at rapportere alle formodede bivirkninger via hjemmesiden: www. Agenziafarmaco.gov .it/it/responsabili fra det italienske lægemiddelagentur.
04.9 Overdosering
Symptomer
I et klinisk studie, der evaluerede den gentagne administration af 4 g strontiumranelat dagligt i mere end 25 dage hos raske postmenopausale kvinder, blev der fundet god tolerabilitet. En enkelt administration af doser op til 11 g hos unge raske mandlige frivillige forårsagede ikke særlige symptomer.
Ledelse
Fra observation af overdosepisoder i kliniske forsøg (op til 4 g / dag i en maksimal tidsperiode på 147 dage) er der ikke observeret klinisk relevante effekter.
Indgivelse af mælk eller antacida kan være nyttig til at reducere absorptionen af den aktive ingrediens.I tilfælde af en betydelig overdosis kan muligheden for at fremkalde opkastning for at eliminere den ikke-absorberede aktive ingrediens overvejes.
05.0 FARMAKOLOGISKE EGENSKABER
05.1 Farmakodynamiske egenskaber
Farmakoterapeutisk gruppe: Lægemidler til behandling af knoglesygdomme - andre lægemidler, der påvirker knoglestruktur og mineralisering.
ATC -kode: M05BX03.
Handlingsmekanisme
In vitro, strontiumranelat:
- øger knogledannelse i knoglevævskulturer samt replikation af osteoblastforstadier og kollagensyntese i knoglecellekulturer.
- reducerer knogleresorption ved at reducere osteoklastdifferentiering og deres resorptionsaktivitet.
Dette bestemmer en genbalancering af knogleomsætning til fordel for dens dannelse.
Strontiumranelats aktivitet er blevet påvist i flere eksperimentelle undersøgelser, især hos intakte rotter øger strontiumranelat den trabekulære knoglemasse, antallet og tykkelsen af trabeculae, hvilket resulterer i en forbedring af knoglestyrke.
Strontium absorberes hovedsageligt på den krystallinske overflade, og kun i begrænset omfang erstatter det calcium i apatitkrystallen i nydannet knogle hos både dyr og mennesker under behandling. Strontiumranelat ændrer ikke knoglekrystalets egenskaber. I iliac crest -knoglebiopsier taget efter behandling med strontiumranelat 2 g / dag i op til 60 måneder i fase III -undersøgelser blev der ikke observeret nogen skadelige virkninger på knoglekvalitet eller mineralisering.
De kombinerede virkninger af fordelingen af strontium i knogler (se afsnit 5.2) og den højere røntgenabsorption af strontium sammenlignet med calcium, fører til en stigning i værdien af knogletæthed (BMD) målt ved dobbeltstråle fotonabsorptiometri ( DXA). Tilgængelige data indikerer, at disse faktorer tegner sig for cirka 50% af de observerede ændringer i BMD over 3 års behandling med PROTELOS 2 g / dag. Dette bør tages i betragtning ved vurdering af ændringer i BMD under behandling med PROTELOS. I fase III -undersøgelser, der demonstrerede effekten af PROTELOS -behandling til reduktion af frakturer, øgede PROTELOS den gennemsnitlige BMD sammenlignet med inklusion med cirka 4% årligt i lændehvirvlerne og 2% årligt i lændehvirvlerne. Af lårhalsen, som når, afhængigt af undersøgelsen, henholdsvis fra 13 til 15% og fra 5 til 6% efter 3 år.
I fase III-undersøgelser, sammenlignet med placebo, blev biokemiske markører for knogledannelse (specifik alkalisk phosphatase og C-terminal propeptid af type I procollagen) øget og knogleresorption (serum C-telopeptid og urin-tværbindinger af N-telopeptid) faldet fra den tredje måned til det tredje behandlingsår.
Ud over de primære farmakologiske virkninger af strontiumranelat er der observeret et lille fald i serumkalcium- og parathyreoideahormon (PTH) niveauer, stigninger i blodfosforkoncentration og total alkalisk fosfataseaktivitet uden kliniske konsekvenser.
Klinisk effekt
Osteoporose er defineret som BMD i rygsøjlen eller hoften, der er 2,5 eller flere standardafvigelser lavere end middelværdien i den unge normale befolkning. Nogle risikofaktorer er forbundet med postmenopausal osteoporose, herunder lav knoglemasse, lav knoglemineraltæthed, tidlig overgangsalder, rygning og en familiehistorie af osteoporose. Den kliniske konsekvens af osteoporose er brud, risikoen for brud stiger i takt med at antallet af risikofaktorer stiger.
Behandling af postmenopausal osteoporose
Studieprogrammet til evaluering af brudreduktion med PROTELOS bestod af to fase III placebokontrollerede undersøgelser: SOTI-undersøgelsen og TROPOS-undersøgelsen. SOTI -undersøgelsen omfattede 1.649 postmenopausale kvinder med dokumenteret osteoporose (lav lumbal BMD og udbredte hvirvelbrud) og en gennemsnitsalder på 70 år. TROPOS -undersøgelsen omfattede 5.091 postmenopausale kvinder med osteoporose (BMD med lav lårhals og mindst et brud hos mere end halvdelen af patienterne) og en gennemsnitsalder på 77 år. Tilsammen indgik SOTI- og TROPOS -undersøgelserne 1.556 patienter over 80 år på inklusionstidspunktet (23,1% af undersøgelsespopulationen). I begge undersøgelser blev patienter ud over terapi (2 g / dag strontium eller placebo), patienter tager tilstrækkelige calcium- og D -vitamintilskud.
PROTELOS reducerede den relative risiko for nye vertebrale frakturer med 41% over 3 års behandling i SOTI -undersøgelsen (tabel 1). Effekten var signifikant fra det første år. Lignende fordele blev påvist hos kvinder med flere frakturer ved indskrivning. Med hensyn til kliniske hvirvelbrud (defineret som frakturer forbundet med rygsmerter og / eller et fald i kropshøjde på mindst 1 cm) blev den relative risiko reduceret med 38%. PROTELOS reducerede også antallet af patienter med reduceret "kropshøjde på mindst 1 cm sammenlignet med placebo.Livskvalitetsvurderingen ved hjælp af den specifikke QUALIOST-skala samt de generelle sundhedsopfattelsesresultater for den generelle SF-36-skala angiver fordelene ved PROTELOS sammenlignet med placebo.
PROTELOS effektivitet til at reducere risikoen for nye vertebrale frakturer blev bekræftet af TROPOS -undersøgelsen, selv for osteoporotiske patienter uden skrøbelighedsbrud, på tidspunktet for inklusion.
En fælles analyse af SOTI- og TROPOS -undersøgelserne viste, at hos patienter over 80 år på inklusionstidspunktet reducerede PROTELOS den relative risiko for nye hvirvelbrud med 32% over 3 års behandling (forekomst af 19, 1% med strontiumranelat vs 26,5% med placebo).
I en analyse i kølvandet af patienter i SOTI- og TROPOS -undersøgelserne med lændehvirvler og / eller lårbenshals BMD i det osteopeniske område på inklusionstidspunktet og uden udbredte frakturer, men med mindst en yderligere frakturrisikofaktor (N = 176), reducerede PROTELOS risikoen af en første hvirvelbrud med 72% over 3 år (forekomst af hvirvelbrud 3,6% med strontiumranelat vs 12,0% med placebo).
En analyse i kølvandet blev udført i en undergruppe af TROPOS-patienter af særlig medicinsk interesse og med høj risiko for brud [defineret som patienter med lårhals BMD T-score ≤-3 SD (producentinterval svarende til -2,4 SD ifølge NHANES III) og en alder ≥ 74 år (n = 1.977, dvs. 40% af TROPOS -undersøgelsespopulationen)]. I denne gruppe, over 3 års behandling, reducerede PROTELOS risikoen for hoftebrud 36% sammenlignet med placebo (tabel 2).
Behandling af osteoporose hos mennesker
Effekten af PROTELOS blev påvist hos mænd med osteoporose i et dobbeltblindet, placebokontrolleret, 2-årigt studie med en hovedanalyse foretaget efter et år hos 243 patienter (befolkning Hensigt at behandle, 161 patienter behandlet med strontiumranelat) med høj risiko for brud (gennemsnitsalder 72,7 år; gennemsnitlig lumbal BMD med en T -score på -2,6; 28% udbredt vertebralfraktur).
Alle patienter modtog daglige tilskud af calcium (1000 mg) og D -vitamin (800 IE).
Statistisk signifikante stigninger i BMD -værdier blev observeret så tidligt som 6 måneder efter behandlingsstart med PROTELOS sammenlignet med placebo.
En statistisk signifikant stigning i de gennemsnitlige BMD-værdier i lændehvirvelsøjlen blev observeret i løbet af 12-månedersperioden, det vigtigste effektkriterium (E (SE) = 5,32%; 95% CI = [3,86; 6,79]: p overgangsalderen.
Der blev observeret statistisk signifikante stigninger i lårhalsens BMD og totale lårbenets BMD -værdier (s
Pædiatrisk population
Det Europæiske Lægemiddelagentur har frafaldet forpligtelsen til at indsende resultaterne af undersøgelser med PROTELOS i alle undergrupper af den pædiatriske population i osteoporose (se afsnit 4.2 for information om pædiatrisk brug).
05.2 "Farmakokinetiske egenskaber
Strontiumranelat består af 2 stabile strontiumatomer og et molekyle ranelsyre, en organisk komponent, der repræsenterer det bedste kompromis med hensyn til molekylvægt, farmakokinetik og accept af lægemidlet. Farmakokinetikken for strontium og ranelsyre blev evalueret hos raske unge mandlige frivillige, hos raske postmenopausale kvinder og under langtidsbehandling hos mænd med osteoporose og hos kvinder med postmenopausal osteoporose, herunder ældre.
Absorption, distribution, binding af ranelsyre med plasmaproteiner er lav på grund af dens høje polaritet. Der er ingen ophobning af ranelsyre og ingen tegn på metabolisme hos dyr og mennesker. Absorberet ranelsyre elimineres hurtigt uændret via urinen.
Absorption
Den absolutte biotilgængelighed af strontium er 25% (område 19-27%) efter en oral dosis på 2 g strontiumranelat. Maksimal plasmakoncentration nås 3-5 timer efter en enkelt dosis på 2 g.
Steady state opnås efter 2 ugers behandling. Indtagelse af strontiumranelat med calcium eller mad reducerer biotilgængeligheden af strontium med cirka 60 - 70%sammenlignet med administration 3 timer efter et måltid På grund af den relativt langsomme absorption af strontium bør fødeindtagelse og calcium før og efter indtagelse af PROTELOS undgås . Oralt vitamin D -tilskud forstyrrer ikke strontiumeksponering.
Fordeling
Strontium har et distributionsvolumen på ca. 1 l / kg. Bindingen af strontium til humane plasmaproteiner er lav (25%), og strontium har en "høj affinitet for knoglevæv. Måling af strontiumkoncentrationer i iliac crest -knoglebiopsier hos patienter behandlet i op til 60 måneder med 2 g / dag strontiumranelat , viser, at koncentrationen af strontium i knogle kan nå et plateau efter cirka 3 års behandling Der er ingen patientdata, der viser kinetikken ved eliminering af strontium fra knogle efter seponering.
Biotransformation
Som en divalent kation metaboliseres strontium ikke. Strontiumranelat hæmmer ikke enzymkomplekset cytochrom P450.
Eliminering
Eliminering af strontium er uafhængig af tid og dosis. Strontiums effektive halveringstid er cirka 60 timer. Strontiumudskillelse sker via nyrerne og mave -tarmkanalen. Plasmaclearance er cirka 12 ml / min (CV 22%) og renal clearance cirka 7 ml / min (CV 28%).
Farmakokinetik i især populationer
Ældre patienter
Befolkningens farmakokinetiske data viste ingen sammenhæng mellem alder og tilsyneladende clearance af strontium i målpopulationen.
Nyresvigt
Hos patienter med moderat til moderat nedsat nyrefunktion (kreatininclearance 30-70 ml / min) falder strontiumclearance, efterhånden som kreatininclearance falder (ca. 30% fald i kreatininclearanceområdet 30 til 70 ml / min); dette fører til en stigning i plasma strontiumniveauer. I fase III -undersøgelserne havde 85% af patienterne en kreatininclearance på mellem 30 og 70 ml / min, 6% mindre end 30 ml / min overhovedet. "inklusion og gennemsnitlig kreatininclearance var 50 ml / min. Derfor , ingen dosisjustering er nødvendig hos patienter med moderat til moderat nedsat nyrefunktion Der er ingen farmakokinetiske data om patienter med svært nedsat nyrefunktion (kreatininclearance
Leverinsufficiens
Der er ingen farmakokinetiske data om patienter med leverinsufficiens. På grund af de farmakokinetiske egenskaber ved strontium forventes der ingen effekt.
05.3 Prækliniske sikkerhedsdata
Ikke-kliniske data afslører ingen særlig fare for mennesker baseret på konventionelle undersøgelser af sikkerhed farmakologi, gentoksicitet, kræftfremkaldende potentiale.
Hos gnavere resulterede kronisk oral administration af høje doser strontiumranelat i knogle- og tandabnormiteter, der hovedsageligt består af spontane frakturer og forsinket mineralisering, reversibel efter afbrydelse af behandlingen. Disse effekter blev set med et niveau af strontium i knoglerne 2 til 3 gange højere end niveauerne set hos mennesker efter behandling, der varede op til 3 år. Data om skeletakkumulering af strontiumranelat ved langvarig eksponering er begrænsede.
Undersøgelser vedrørende udviklingstoksicitet har resulteret i abnormiteter i knogler og tænder hos rotter og kaniner (f.eks. Bukning af lange knogler og bølgede ribben). Disse virkninger er reversible 8 uger efter behandlingens ophør.
Miljørisikovurdering (ERA)
Miljørisikovurderingen af strontiumranelat blev udført i overensstemmelse med de europæiske retningslinjer vedrørende ERA.
Strontiumranelat udgør ikke en risiko for miljøet.
06.0 LÆGEMIDDELOPLYSNINGER
06.1 Hjælpestoffer
Aspartam (E 951)
Maltodextrin
Mannitol (E 421)
06.2 Uforenelighed
Ikke relevant.
06.3 Gyldighedsperiode
- 3 år.
- Efter rekonstituering i vand er suspensionen stabil i 24 timer. Det anbefales dog at drikke suspensionen umiddelbart efter tilberedning (se pkt.4.2).
06.4 Særlige opbevaringsforhold
Denne medicin kræver ingen særlige opbevaringsbetingelser.
Opbevaringsbetingelser efter rekonstituering af lægemidlet, se afsnit 6.3.
06.5 Den umiddelbare emballages art og emballagens indhold
Papir / polyethylen / aluminium / polyethylenposer.
Pakker
Pakninger med 7, 14, 28, 56, 84 eller 100 breve.
Ikke alle pakningsstørrelser er nødvendigvis markedsført.
06.6 Brugsanvisning og håndtering
Ingen særlige instruktioner.
07.0 INDEHAVER AF MARKEDSFØRINGSTILLADELSE
LES LABORATOIRES SERVIER
50, rue Carnot
92284 Suresnes cedex
Frankrig
08.0 MARKEDSFØRINGSTILLADELSESNUMMER
EU/1/04/288/003
A.I.C. n ° 036558031 / E - pakke med 28 breve
09.0 DATO FOR FØRSTE TILLADELSE ELLER FORNYELSE AF TILLADELSEN
Dato for første godkendelse: 21. september 2004
Dato for seneste fornyelse: 22. maj 2014
10.0 DATO FOR REVISION AF TEKSTEN
06/2014