Almindelighed
Crouzons syndrom er en sjælden genetisk sygdom, som bestemmer tilstedeværelsen af kraniosynostose og andre temmelig ejendommelige ansigtsanomalier.
Dens udseende skyldes visse ændringer i DNA'et, der udgør FGFR2- og FGFR3 -generne; disse genetiske elementer er involveret i knoglemodningsprocessen under embryonal udvikling.

Terapien består af en række kirurgiske indgreb, der sigter mod at løse de vigtigste og farligste symptomer.
I øjeblikket har prognosen tendens til at være meget ofte positiv.
Gennemgang af genetik
Inden vi fortsætter med beskrivelsen af Crouzon syndrom, er det nyttigt at gennemgå nogle grundlæggende genetiske begreber.
Hvad er DNA? Det er den genetiske arv, hvor de somatiske træk, forudsætningerne, de fysiske kvaliteter, karakteren osv. Af en levende organisme er skrevet. Den er indeholdt i alle celler i kroppen, der har en kerne, som ligger inden for dette.
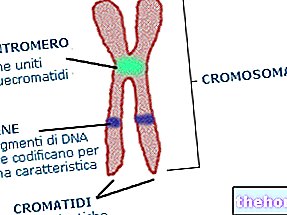
Hvad er kromosomer? Ifølge definitionen er kromosomer de strukturelle enheder, hvor DNA er organiseret. Menneskelige celler indeholder i deres kerne 23 par homologe kromosomer (22 af den autosomale ikke-seksuelle type og et par af den seksuelle type); hvert par er forskelligt fra et andet, da det indeholder en specifik gensekvens.
Hvad er gener? De er korte strækninger eller sekvenser af DNA med en grundlæggende biologisk betydning: fra dem stammer faktisk proteiner eller biologiske molekyler, der er grundlæggende for livet. I generne er der "skrevet" en del af, hvem vi er, og hvem vi bliver.
Hvert gen findes i to versioner, allelerne: en allel er af moderlig oprindelse, derfor overført af moderen; den anden allel er af faderlig oprindelse, derfor overført af faderen.
Hvad er en genetisk mutation? Det er en fejl i DNA -sekvensen, som danner et gen. På grund af denne fejl er det resulterende protein enten defekt eller helt fraværende.I begge tilfælde kan virkningerne være skadelige både for cellens liv, hvor mutationen forekommer, og for organismen som helhed. Medfødte sygdomme og neoplasmer (dvs. tumorer) tilhører en eller flere genetiske mutationer.
Det samme er Crouzons syndrom
Crouzon syndrom er en sjælden genetisk tilstand præget af kraniosynostose og "unaturlig udvikling af visse ansigtselementer, herunder øjne, næse, kæbe og kæbe."
Det er en medfødt sygdom, hvis typiske egenskaber kan være tydelige allerede i de første øjeblikke af livet.
BETYDNING AF CRANIOSINOSTOSI
Kraniosynostose er det udtryk, hvormed læger henviser til den for tidlige fusion af en eller flere kranielle suturer.

Fra webstedet: thecraniofacialcenter.com
Kranialsuturer er de fibrøse led, der forbinder knoglerne i kranialhvelvet sammen (dvs. de frontale, tidsmæssige, parietale og occipitale knogler).
Under normale forhold sker fusion af kranialsuturerne i postnatalperioden (nogle processer slutter endda i en alder af 20 år). Denne lange fusionsproces gør det muligt for hjernen at vokse og udvikle sig tilstrækkeligt.
Hvis fusionen, som i tilfælde af kraniosynostoser, finder sted for tidligt - derfor under prænatal, perinatal * eller tidligt barndom - undergår hjernens elementer (hjerne, lillehjerne og hjernestamme) og nogle sanseorganer (især øjne) en "ændring" af form og vækst.
* Perinatalbetegnelsen refererer til den levetid, der går fra den 27. drægtighedsuge til de første 28 dage efter fødslen.
Oprindelse af navnet
Crouzon syndrom skylder sit navn til den franske læge Octave Crouzon, der har fortjenesten først at have beskrevet sine vigtigste kliniske egenskaber.
Crouzon levede mellem slutningen af 1800'erne og begyndelsen af 1900'erne, præcis fra 1874 til 1938. Oprindeligt brugte han udtrykket kraniofacial dysostose for at definere det syndrom, der senere tog hans navn.
Årsager
Crouzon syndrom opstår som et resultat af en mutation i FGFR2 -genet, der er placeret på kromosom 10, eller FGFR3 -genet, der er placeret på kromosom 4.
FGFR er det engelske akronym for Fibroblast -vækstfaktorreceptor, der oversættes til italiensk er: Receptor for fibroblastvækstfaktoren.
Den funktionelle rolle for FGFR2- og FGFR3 -generne er hver at producere et receptorprotein, som igen har til opgave at regulere modning og embryonisk udvikling af knoglevæv.
Ifølge forskernes teorier ville mutationerne i FGFR2 og FGFR3 hyperstimulere de samme gener, hvilket, igen en gang aktiv, ville fremkalde en tidlig modning af nogle knoglevæv, herunder dem, der udgør kraniet.
GENETIK
De genetiske mutationer, der er ansvarlige for Crouzon syndrom, kan være arvelige eller kan opstå spontant efter undfangelsen.
I det første tilfælde har den sygelige tilstand - som læger også omtaler som arvelig Crouzon syndrom - alle egenskaber ved en autosomal dominerende genetisk sygdom (eller arvelig dominerende sygdom). For en ny læser af genetik betyder det, at:
- Sygdommen og dens symptomer forekommer også i nærvær af kun en muteret genallel (det er ligegyldigt om den kommer fra moderen eller faderen), da sidstnævnte er dominerende over den raske.
- En forælder, der bærer mutationen, er nok til at have sygdommen i en del af afkommet.
- Sandsynligheden for, at et sygt barn vil blive født, fra et par, hvor kun en af de to komponenter bærer mutationen, er 50%.
I det andet tilfælde er den sygelige tilstand - som eksperter angiver med terminologien for ikke -arveligt Crouzon -syndrom - imidlertid et resultat af en anomal sporadisk hændelse, som ændrer DNA'et under fosterets embryonale vækst.
Resumé af betydningen af udtrykkene arvelig, autosomal og dominerende
- Arvelig: det betyder, at forældrene overfører den genetiske ændring, der er ansvarlig for sygdommen, til afkommet (dvs. til børnene).
- Autosomal: det betyder, at mutationen, der er ansvarlig for sygdommen, befinder sig i et ikke-kønskromosom, derfor autosomalt.
- Dominant: betyder, at sygdommen forårsager symptomer og tegn, selv når kun en allel af det ansvarlige gen er muteret. I enklere termer er det som om allelen med mutationen havde mere magt end den sunde allel.
EPIDEMIOLOGI
Ifølge nogle estimater af forekomsten af Crouzon syndrom ville omkring en ud af 60.000 børn blive født med denne sjældne tilstand.
Crouzon syndrom tegner sig for 4,5% af tilfælde af kraniosynostose.
Symptomer og komplikationer
Patienter med Crouzon syndrom har et meget specifikt symptombillede, som normalt består af:
- Problemer relateret til kraniosynostose, herunder:
-

Fra https://da.wikipedia.org/wiki/Plagiocephaly Brachycephaly, som er klemningen af bagsiden af hovedet. For tidlig fusion af de koronale kranialsuturer følger (koronal kraniosynostose).
Hvis det ikke behandles, kan det påvirke hjernens vækst og udvikling af kognitive evner.
De repræsenterer et "alternativ til brachycephaly: trigonocephaly (fusion af metopisk sutur), dolichocephaly (fusion af sagittal sutur) og plagiocephaly (fusion af koronale suturer). - Exophthalmos, som er betegnelsen for øjenkuglernes fremspring. Det kan betyde tilstedeværelse af synsproblemer.
- Okulær hypertelorisme, det vil sige øjne, der er overdrevet fjernt fra hinanden. Med exophthalmos kan det gøre synsproblemer værre.
- Deformeret næse, generelt i form af et næb. Hvis alvorlig eller ikke behandles kirurgisk, kan denne abnormitet føre til vejrtrækningsproblemer eller de samme symptomer som obstruktiv søvnapnø syndrom.
- Øget intrakranielt tryk. Det er også kendt som intrakraniel hypertension. Dens tilstedeværelse forklares ved, at hjernestrukturer ikke har det rigtige rum til at vokse.
Normalt findes i midten af sen barndom, intrakraniel hypertension er en potentiel årsag til hovedpine, opkastning og øjenpine. - Hydrocephalus, som er resultatet af en stigning i cerebrospinalvæsken i det subaraknoidale rum og i cerebrale ventrikler.
- Arnold-Chiari misdannelse (eller Arnold-Chiari syndrom). Det er en deformitet placeret i bunden af kraniet.
* Hydrocephalus og Arnold-Chiari misdannelse er generelt to komplikationer, der opstår i mangel af passende behandlinger. - Abnormiteter i underkæben og maxilla.
Den første har mindre dimensioner end normalt, mens den anden har en tendens til at stikke udad. Alt dette ændrer ganens form og tandstilladset (fravær af nogle tænder osv.), Med konsekvenser (nogle gange endda alvorlige) på fonation og på tygge.
Nogle patienter er født med læbespalte (ganespalte) eller ganespalte.
- Høreproblemer.
55% af patienterne med Crouzon syndrom er født uden øregange eller med store abnormiteter i dem. Dette resulterer i en fraværende eller stærkt reduceret akustisk kapacitet.
Nogle emner udvikler et sæt høreproblemer i voksenalderen, der kan tilskrives det typiske kliniske billede af Ménières syndrom.
- Ledsmerter i nakken.
De vedrører 30% af tilfældene med Crouzon syndrom.
- Hudafvigelser.
Patienter med muteret FGFR3-understøttet Crouzon syndrom er til stede acanthosis nigricans, en dermatose karakteriseret ved en stigning i tykkelse (hyperkeratose) og mørkere (hyperpigmentering) af huden.

To andre anatomiske anomalier forbundet (omend sjældent) med Crouzon syndrom
- Patent arteriel kanal
- Koartation af aorta
CROUZON SYNDROME OG IQ
Takket være de nuværende muligheder for behandling af kraniosynostose har i dag 97% af patienterne med Crouzon syndrom "normal intelligens".
Diagnose
En erfaren børnelæge kan muligvis diagnosticere Crouzon syndrom alene ved hjælp af fysisk undersøgelse.
I nærvær af tvivl eller forvirring er følgende grundlæggende for at nå frem til en præcis konklusion:
- Radiologiske billeder, leveret af røntgenstråler eller CT-scanninger af hovedet
- En gentest, der sigter mod at lede efter eventuelle DNA -mutationer.
MÅL EKSAMEN
Den fysiske undersøgelse består i en nøjagtig analyse af hovedet og de anomalier, der findes på det.
Kranial deformiteter, forårsaget af kraniosynostose (for eksempel brachycephaly), er blandt de mest karakteristiske kliniske tegn på Crouzon syndrom, og som lægen baserer en del af sine diagnostiske konklusioner på.
RADIOLOGISKE UNDERSØGELSER
Røntgen- og CT-scanninger af hovedet viser, hvilke kranialsuturer der har smeltet for tidligt.
Den kraniosynostose, der kendetegner Crouzons syndrom, påvirker de koronale suturer, derfor er en fundet fusion på niveau med sidstnævnte en meget ofte afgørende information til diagnostiske formål.
GENETISK Undersøgelse
Ud over at vise, om DNA'et har mutationer, hjælper genetisk test med at identificere det nøjagtige gen, der forårsager Crouzon syndrom, uanset om det er FGFR2 eller FGFR3.
Behandling
I dag kan bærere af Crouzon syndrom regne med forskellige behandlinger afhængigt af sværhedsgraden af tilstanden og symptomerne.
Faktisk har lægerne sikret:
- Kirurgi til løsning af kraniosynostose og dens symptomer.
- Akustiske hjælpemidler, i tilfælde af høreproblemer.
- Terapier til forbedring af sprogkundskaber.
- Kirurgiske terapier til forbedring af anomalier i overkæben og underkæben.
- En operation, kendt som en trakeostomi, for at løse vejrtrækningsproblemer.
Bemærk venligst: Crouzons syndrom er en sygelig tilstand, der stammer fra en "genetisk ændring af DNA'et, der er umuligt at helbrede. Så faktisk behandler læger kun sygdommen fra et symptomatologisk synspunkt.
KIRURGI TIL CRANIOSYNOSTOSE
De terapeutiske formål med den kirurgiske operation er to:
- Giv hjernens strukturer og øjne den plads, de har brug for til at udvikle og fungere bedst muligt.
- Giv hovedet en normal form, og løs derefter problemet med brachycephaly.
Kirurger har mulighed for at udføre operationen på to forskellige måder (eller tilgange): gennem en "traditionel kirurgisk operation - også kaldet" åben " - eller gennem en" endoskopisk operation.
Den "åbne operation" involverer "udførelsen af et" snit på hovedet, hvorigennem den opererende læge udtrækker knoglen eller de misdannede kranieknogler, som skal ombygges. Ved afslutningen af ombygningen genindsætter kirurgen knoglestrukturer, der tidligere blev ekstraheret, og lukker snittet med suturer.
Den endoskopiske operation involverer derimod brug af et endoskop og øvelse af et meget lille snit på hovedet, hvorigennem den opererende læge indsætter selve endoskopet.
Endoskopet er faktisk et tyndt og fleksibelt rør, udstyret med et fiberoptisk kamera (for enden indsat i kraniet) og forbundet til en skærm. Gennem dette særlige instrument og de billeder, det projekterer på skærmen, er kirurgen i stand til at adskille de fusionerende kranialsuturer for tidligt, med bemærkelsesværdig præcision og uden at ty til hudindsnit og knogleekstraktioner.
Ifølge eksperter er det bedste tidspunkt at udføre operationen i meget tidlig barndom (første 12 måneder af livet), da knoglerne lettere formes.
Det skal dog huskes, at jo yngre patienten er, desto større er risikoen for gentagelse af de samme kranielle suturer (gentagelse) I tilfælde af gentagelse skal operationen gentages.
Ifølge nogle statistiske undersøgelser skal 10-20% af meget unge forsøgspersoner, der gennemgår kraniosynostoseoperation, gennemgå en anden operation på grund af et tilbagefald.
BEHANDLING AF AKUSTISKE PROBLEMER
Udover at ordinere brugen af høreapparater, anbefaler læger også periodisk auditiv kontrol, da dette er den bedste måde at forhindre forværring af eksisterende problemer.
KIRURGISKE TERAPIER FOR KÆBE- OG KÆBEANOMALIER
Behandlingen af kæbe- og underkæbe -anomalier omfatter kirurgi for justering af kæben og / eller underkæben, nogle tandbehandlinger til arrangement af tandbuerne og operationen for opløsning af læbe- og / eller ganespalte.
TRACHEOSTOMI
Trakeostomi er den kirurgiske operation, hvorigennem lægen på halsniveauet (hvor luftrøret passerer) skaber en passage for luft bestemt til lungerne. Dette giver dem, der gennemgår denne operation, mulighed for at trække vejret igen og korrekt.
For at transportere luften ind i lungerne har du brug for et lille rør, kaldet et transcheostomirør, som er den rigtige størrelse til indsættelse i luftrøret.
Prognose
Generelt afhænger prognosen af sværhedsgraden af kraniosynostosen: hvis sidstnævnte kan behandles med gode resultater, kan patienter med Crouzon syndrom nyde et næsten normalt liv.